ВВЕДЕНИЕ
Сточные воды процесса производства тринитротолуола (ТНТ), окрашенные в красный цвет, содержат сульфит натрия, который может быть выделен из раствора. Согласно процессу, разработанному В.Р. Куком, эти сточные воды подвергают обработке для выделения соединений натрия без рецикла золы, образующейся при сжигании, и без формования из нее твердых гранул.
Глава 1. ПРОМЫШЛЕННЫЕ СПОСОБЫ ПОЛУЧЕНИЯ ОСНОВНЫХ ПРОИЗВОДНЫХ БЕНЗОЛА
Получение алкилпроизводных бензола
В промышленности при синтезе алкилпроизводных бензола в качестве алкилирующих агентов применяют главным образом алкилгалогениды (в основном хлорпроизводные) и олефины. В качестве катализатора при алкилировании алкилгалогенидами используют только хлорид алюминия, отличающийся наибольшей активностью из всех доступных апротых кислот. Он же может применяться при алкилировании бензола олефинами, но в этом случае пригодны и другие катализаторы кислотного типа (H2SO4, безводный HF, BF3, фосфорная кислота на носителях, алюмосиликаты, цеолиты).
Использование спиртов в качестве алкилирующих агентов менее эффективно, потому что при алкилировании спиртами АlСl3 разлагается, а протые кислоты разбавляются образующейся водой. В обоих случаях происходит дезактивирование катализатора, что обусловливает его большой расход.
При реакции с хлорпроизводными или олефинами АlСl3 расходуется только в каталитических количествах. В первом случае он активирует атом хлора, образуя сильно поляризованный комплекс или ион карбония (уравнение (4.1.10)), что с олефинами происходит только в присутствии сокатализатора — хлороводорода (уравнение (4.1.11)).
В твердом виде хлорид алюминия практически нерастворим в бензоле и является слабым катализатором. Однако в присутствии хлороводорода хлорид алюминия начинает превращаться в темное жидкое вещество, так называемый комплекс Густавсона, обладающий высокой каталитической активностью, и реакция алкилирования постепенно ускоряется. Комплекс Густавсона можно приготовить, пропуская HCl при нагревании через суспензию АlСl3 в ароматическом углеводороде. Комплекс представляет собой соединение АlСl3 и HCl с 1–6 молекулами ароматического углеводорода, одна из которых находится в особом структурном состоянии положительно заряженного иона (σ-комплекс), а остальные образуют сольватную оболочку:

Во избежание медленного катализа твердым хлоридом алюминия этот активный каталитический комплекс целесообразно готовить предварительно и потом подавать в реакцию. Кроме HCl его образованию способствуют небольшие добавки воды или соответствующего хлорпроизводного, роль которых состоит в генерации HCl. Более приемлемо использовать HCl или RCl, т. к. вода дезактивирует часть катализатора, разлагая его. По этой же причине необходимо хорошо осушать реагенты и следить, чтобы в реакционную смесь не попадала вода, способная вызвать бурное разложение комплекса. Другими катализаторными ядами являются многие сернистые соединения и аммиак, в меньшей степени — диены и ацетилен. Следовательно, жидкая реакционная масса при алкилировании с хлоридом алюминия состоит из двух фаз: каталитического комплекса и углеводородного слоя.
При использовании в качестве катализаторов:
H2SO4 и HF процесс алкилирования проводят в жидкой фазе при 10–40°С и давлении 0,1–1 МПа;
Н3РО4 — в газовой фазе при 225–275 °С и 2–6 МПа;
алюмосиликатов и цеолитов — в жидкой или газовой фазе при 200–400°С и том же давлении.
В недавнем прошлом широко применялся при алкилировании твердый фосфорно-кислотный катализатор, в настоящее время больше внимание уделяется цеолитам, но преобладающее промышленное значение все же имеет хлорид алюминия, обладающий перед другими катализаторами рядам существенных преимуществ.
Из алкилпроизводных бензола важнейшее практическое значение имеют этилбензол (8) и кумол [(9); изопропилбензол]. Их получают сотнями тысяч т при действии на бензол этилена или пропилена соответственно в присутствии хлорида алюминия и хлороводорода (уравнение (4.1.13)).
Синтез ведут в аппаратах непрерывного действия, представляющих собой эмалированные или футерованные графитовой плиткой многосекционные колонны. Хлорид алюминия вводится в аппараты в виде заранее приготовленного раствора, содержащего 10–12 % AlCl3, 50–60 % бензола и 25–30 % соответствующего диалкилбензола, т. к. в бензоле AlCl3 растворяется плохо. Для образования хлороводорода, который является сокатализатором, в раствор добавляют воду (2 % от массы AlCl3). В нижнюю часть колонны через распылитель подается алкен.
Так как этилбензол (8) и кумол (9) алкилируются быстрее бензола, в процессе реакции неизбежно образование диалкилбензолов (преимущественно мета- и пара-изомеров). Основным фактором, влияющим на соотношение образующихся продуктов, является степень конверсии («глубина алкилирования»). Чтобы предотвратить образование нежелательных диалкилбензолов, глубину алкилирования в этих процессах поддерживают на уровне 10 %. Однако наряду с целевыми продуктами образуются заметные количества диалкилпроизводных, которые отделяются при ректификации; не вступивший в реакцию бензол возвращается в реактор.
На рис. 1 изображена технологическая схема производства этил- или изопропилбензола алкилированием бензола газообразным олефином в присутствии AlCl3.
Свежий бензол вместе с бензолом, возвращенным со стадии разделения, поступает в колонну 3, предназначенную для осушки бензола азеотропной ректификацией. Низкокипящая азеотропная смесь бензола с водой конденсируется в конденсаторе 4 и разделяется в сепараторе 5 на два слоя. Воду с растворенным в ней бензолом отводят (ее можно использовать для промывки реакционной массы), а бензольный слой стекает на верхнюю тарелку колонны 3, создавая орошение. Осушенный бензол из куба колонны 3 в теплообменнике 2 подогревает бензол, идущий на осушку, и попадает в сборник 8, откуда насосом непрерывно закачивается в алкилатор 9.
Каталитический комплекс готовят в аппарате 6 с мешалкой и рубашкой для обогрева паром. В него загружают полиалкилбензолы (ПАБ) или смесь бензола и полиалкилбензола (примерно в отношении 1 : 1) и хлорид алюминия (1 моль на 2,5–3 моль ароматических углеводородов), после чего при нагревании и перемешивании подают хлорпроизводное. Приготовленный комплекс периодически вводят в алкилатор.
Реакция проводится в непрерывно действующей колонне-алкилаторе 9 с горячим сепаратором 12 для отделения каталитического комплекса и обратным конденсатором 10 для возвращения испарившегося бензола и отвода тепла. Олефин поступает в низ колонны, предварительно проходя расходомер. Бензол из емкости 8 поступает в низ алкилатора, как и конденсат из обратного холодильника 10.

Рис. 1. Технологическая схема производства этилбензола или изопропилбензола: 1 — насосы; 2 — теплообменник; 3 — колонна осушки бензола; 4, 10 — конденсаторы; 5 — сепаратор; 6 — аппарат для получения каталитического комплекса; 7 — кипятильник; 8 — сборник; 9 — алкилатор; 11 — газоотделитель; 12, 16 — сепараторы; 13 — абсорбер; 14 — водяной скруббер; 15 — холодильник; 17, 18 — промывные колонны
Газы, отходящие после конденсатора 10, содержат значительное количество паров легколетучего бензола (особенно при использовании разбавленных фракций олефинов). Для улавливания бензола эти газы направляют в абсорбер 13, который орошается полиалкилбензолами, выделенными из реакционной массы на стадии разделения. Собирающийся в нижней части абсорбера раствор бензола в полиалкилбензолах поступает в реакционный аппарат 9 для переалкилирования. Газы после абсорбера 13 промывают водой в скруббере 14 для удаления НСl и с6расывают в атмосферу или используют в качестве топочного газа.
Углеводородный слой, отбираемый после сепаратора 12, состоит из бензола, моно- и полиалкилбензолов. В нем присутствуют также в небольшом количестве другие гомологи бензола, получившиеся за счет примесей олефинов в исходной фракции или путем частичной деструкции алкильной группы под действием АlСl3.
При синтезе этил- и изопропилбензола реакционная масса содержит: 45–55 масс. % бензола, 35–40 масс. % моноалкилбензола, 8–12 масс. % диалкилбензола и до 3 масс. % более высокоалкилированных соединений, побочных продуктов и смол. Вся эта смесь проходит водяной холодильник 15 и дополнительно отстаивается в холодном сепараторе 16, откуда каталитический комплекс периодически возвращают в алкилатор. Алкилат направляют после этого на очистку от растворенного хлороводорода и следов хлорида алюминия. С этой целью смесь промывают в системе противоточных колонн 17 и 18 вначале водой, а затем водной щелочью. Нейтрализованная смесь углеводородов (алкилат) поступает на ректификацию.
Продукты реакции разделяют в нескольких непрерывно действующих ректификационных колоннах (на схеме не показаны). В первой колонне отгоняют бензол и воду, растворившуюся в углеводородах на стадии промывки. В следующей колонне в вакууме отгоняют фракцию, содержащую главным образом целевой продукт, но с примесью ближайших гомологов бензола. Затем ее подвергают дополнительной ректификации с выделением технического этил- или изопропилбензола. Кубовая жидкость второй колонны содержит полиалкилбензолы с примесью продуктов осмоления, которые образуются под действием АlСl3. Полиалкилбензолы отгоняют в вакууме от смол и используют для абсорбции бензола из отходящих газов и приготовления каталитического комплекса. Через эти промежуточные операции полиалкилбензолы снова возвращают в аппарат 9, где их подвергают деалкилированию. Выход целевого продукта с учетом всех потерь достигает 94–95 % при расходе 10 кг АlСl3 на 1 т моноалкилбензола.
Данная технология алкилирования бензола имеет ряд недостатков и в последнее время непрерывно совершенствуется. Так, для уменьшения количества сточных вод предлагалось разлагать кислотный алкилат небольшим количеством воды, при этом получается концентрированный раствор гексагидрата АlСl3, находящий разнообразное применение. Предлагалось проводить неодинаковые по скорости процессы алкилирования бензола и переалкилирования полиалкилбензолов в разных аппаратах, что снижает количество рециркулята и энергетические затраты и позволяет работать при меньшем избытке бензола по отношению к олефину.
Один из вариантов усовершенствованного процесса алкилирования состоит в применении небольшого количества каталитического комплекса, растворяющегося в алкилате (гомогенное алкилирование). В этом случае, ввиду отсутствия больших масс катализатора, проводят реакцию при 160–200 °С и соответствующем давлении, необходимом для поддержания смеси в жидком состоянии. Схема гомогенного алкилирования бензола представлена на рис. 2.
В алкилатор 1 подают этилен, бензол и небольшое количество каталитического комплекса, снимая выделяющееся тепло кипящим водным конденсатом и генерируя технологический пар (при обычной технологии это тепло не утилизируется). Полученный алкилат поступает в переалкилатор 2, куда подают полиалкилбензолы (ПАБ) со стадии разделения; взаимодействуя с бензолом, они образуют дополнительное количество целевого продукта. Алкилат из аппарата 2 дросселируют до атмосферного давления и подают в сепаратор 3, при этом выделяющуюся энергию полезно утилизируют для испарения части бензола, который конденсируют и возвращают на алкилирование. Жидкий алкилат из сепаратора 3 охлаждают и направляют на нейтрализацию и последующее разделение. По этой технологии уже работают несколько установок большой единичной мощности.

Рис. 2. Технологическая схема процесса гомогенного алкилирования бензола: 1 — алкилатор; 2 — переалкилатор; 3 — сепаратор; 4 — конденсатор; 5 — холодильник
Гидрирование бензола
При гидрировании бензола получают циклогексан (2) — один из трех основных продуктов, определяющих спрос на бензол. Условия гидрирования бензола зависят от степени его чистоты. Гидрирование бензола, очищенного от тиофена, проводят при 140–200 °С и давлении 1–5 МПа; катализатором является никель, нанесенный на оксид хрома(III) или алюминия. При наличии в бензоле примесей, содержащих серу, в качестве катализатора используют сульфиды никеля, кобальта, молибдена или вольфрама, нечувствительные к сернистым соединениям. Эти катализаторы требуют значительно более жестких условий гидрирования: процесс ведут при температуре 320–360 °С и давлении ~30 МПа.
В качестве сырья используют нефтяной или каменноугольный бензол. При использовании нефтяного бензола циклогексан получается высокого качества. Однако в последнее время все чаще используется каменноугольный бензол, который характеризуется повышенным содержанием примесей, и циклогексан, полученный из каменноугольного бензола, нуждается в дополнительной очистке.
На рис. 3 приведена схема процесса производства циклогексана, разработанного Французским институтом нефти. Циклогексан получается высокой степени чистоты. Процесс гидрирования протекает в две стадии: на первой происходит гидрирование основной части бензола на суспендированном непирофорном никеле Ренея, на второй — догидрирование на стационарном катализаторе Ni / Al2O3.
Бензол и водород поступают в основной реактор жидкофазного гидрирования 1, в который предварительно насосом в виде суспензии подается катализатор. Однородность распределения катализатора обеспечивается барботированием газа через жидкость и интенсивной циркуляцией реакционного раствора через выносной теплообменник 3, в котором генерируется технический пар низкого давления. Температура в реакторе регулируется за счет испарения циклогексана. Гидрирование проводится при 200 °С и давлении 4 МПа (парциальное давление водорода ~0,3 МПа).
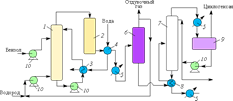
Рис. 3. Технологическая схема процесса производства циклогексана: 1 — основной реактор гидрирования; 2 — вспомогательный реактор гидрирования; 3 — 5, 8 — теплообменники; 6 — сепаратор высокого давления; 7 — колонна стабилизации; 9 — сепаратор; 10 — насосы
Продукты реакции из верхней части основного реактора гидрирования поступают во вспомогательный реактор 2, в котором реализуется практически 100%-я степень превращения бензола в циклогексан. Затем продукты через теплообменники отводятся в сепаратор высокого давления 6 и подвергаются фракционированию в колонне стабилизации 7. Газообразные продукты из сепаратора частично возвращаются на рецикл, а из колонны стабилизации через холодильник 8 поступают в сепаратор 9, из которого отводят готовый продукт. Газы отдувки из сепаратора и колонны стабилизации используются в качестве топлива.
Расход суспендированного катализатора составляет 1 кг на 2,3 т циклогексана. Активность катализатора зависит от содержания оксида углерода в водороде — максимально 0,002 масс. %. Длительность работы катализатора обусловливается содержанием серы в бензоле, которой должно быть не более 0,0001 масс. %. При нормальном режиме установка работает 5–6 месяцев до полной смены катализатора.
Лимитирующими факторами производительности реактора гидрирования являются мощность жидкостных насосов и предельно допустимая скорость барботирования газа, при превышении которой начинается унос жидкости и катализатора с отходящими парами. При использовании бензола 99,9%-й чистоты и насыщенного водой водорода при давлении 3,0–3,4 МПа выход циклогексана практически стехиометрический — 99 %, чистота ≥ 99,8 масс. %.
Окисление бензола
При глубоком окислении бензола происходит деструкция ароматического кольца и образуется малеиновый ангидрид (45). Предполагается, что процесс окисления идет через образование промежуточного продукта — 1,4-бензохинона.
В промышленности этот способ получения малеинового ангидрида используется в больших масштабах. Процесс ведут путем контактно-каталитического окисления бензола в паровой фазе кислородом воздуха, катализаторами являются модифицированные смеси оксидов ванадия и молибдена. Модифицирующими добавками служат соли кобальта, никеля, фосфора, натрия, вольфрама, титана и т. д. Каталитическая масса формуется в гранулы или наносится на носитель, в качестве которого чаще всего используется α-Al2O3. Окисление в этих условиях идет благодаря активации кислорода, хемосорбированного поверхностью катализатора с последующим его взаимодействием с углеводородом.
Смесь бензола с воздухом подогревается контактными газами в теплообменнике 1 до 120–150 °С и поступает в реактор 4 — аппарат, содержащий большое количество реакционных трубок, в которых находится катализатор. Окисление происходит при температуре 370–450 °С. Для отвода тепла реакции в межтрубном пространстве циркулируют расплавы нитрита натрия и нитрата калия. Нагретые соли, в свою очередь, отдают тепло воде, образуя пар высокого давления. Бензол в реакторе окисляется практически полностью.
Контактные газы, выходящие из реактора 4, поступают в теплообменник 1, затем вхолодильник 2, в котором охлаждаются водой до 160–170 °С, после чего направляются в сепаратор 5, где из них выделяется часть малеинового ангидрида. Затем газы через сепаратор 5 поступают в скруббер 6, в котором улавливается водой оставшийся малеиновый ангидрид и другие растворимые в воде продукты реакции; малеиновый ангидрид при этом растворяется в воде, образуя малеиновую кислоту. Выходящий газ выбрасывается в атмосферу. Полученный 40%-й раствор малеиновой кислоты проходит стадию дегидратации в аппарате 7 и вместе с малеиновым ангидридом, выделенным из сепаратора 5, подвергается химической очистке и вакуумной ректификации в колонне 9. Выход малеинового ангидрида составляет 68–72 %.
В настоящее время более совершенными считаются конвертеры с гранулированным катализатором в псевдоожиженном слое. В них эффективно работает весь катализатор, и создаются условия, позволяющие значительно легче отводить тепло и точнее поддерживать необходимую температуру. Это повышает выход продукта на стадии контактирования.
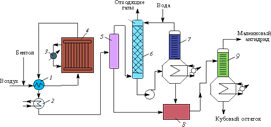
Рис. 4. Технологическая схема производства малеинового ангидрида: 1 — теплообменник; 2 — холодильник; 3 — котел-утилизатор; 4 — контактный аппарат; 5 — сепаратор; 6 — скруббер; 7 — дегидрататор; 8 — емкость для малеинового ангидрида-сырца; 9 — ректификационная колонна
Получение хлорпроизводных бензола
Из хлорпроизводных бензола в промышленности в больших масштабах получают хлорбензол (10) и гексахлорбензол (20).
Промышленное производство хлорбензола — это крупнотажный непрерывный процесс. В качестве катализатора используют хлорид железа(III), получающийся при взаимодействии хлора и металлического железа, загружаемого в аппарат в виде обрезков листового железа. Образующийся безводный хлорид железа(III) растворяется в бензоле и продуктах его хлорирования, благодаря чему реакция идет как гомогенно-каталитическая. Очень важно применять осушенные бензол и хлор, т. к. гидратированный хлорид железа(III) нерастворим в органической фазе и реакция может приобрести менее выгодный гетерогенно-каталитический характер. Кроме того, образующийся при этом хлороводород, растворяясь в воде (соляная кислота), агрессивно действует на металлы.
По мере образования хлорбензола начинается дальнейшее его хлорирование, и неизбежно получаются полихлориды, причем скорость этого процесса растет с повышением концентрации хлорбензола. Чтобы уменьшить количество нежелательных продуктов, реакцию приходится вести с «обратным» бензолом, т. е. обрывать ее задолго до полного израсходования бензола. Например, при периодическом способе производства реакцию заканчивают, когда в смеси содержится около 50 % бензола, 30–40 % хлорбензола и 20–30 % полихлоридов. Использование более прогрессивного непрерывного процесса хлорирования позволяет сократить образование полихлорпроизводных до 1,5–4 %, при этом в реакционной массе остается до 65 % бензола.
В настоящее время в промышленности хлорбензол получают непрерывным методом (рис. 4.1.15) в специальных аппаратах — хлораторах, представляющих собой трубу, заполненную перемешанными стальными и керамическими кольцами. В верхней части хлоратора имеется сепарационный объем, не заполненный насадкой. Бензол и хлор подают в нижнюю часть аппарата. Подачу реагентов регулируют таким образом, чтобы полностью использовать хлор, и чтобы температура в аппарате поддерживалась за счет теплоты реакции (32 кДж/моль) на уровне 76–83 °С. Пары бензола и, в небольшой степени, хлорбензола вместе с выделяющимся хлороводородом из верхней части аппарата поступают в теплообменник, конденсируются, отделяются от хлороводорода и после осушки возвращаются в процесс. Хлороводород направляется на абсорбцию. Реакционная масса, вытекающая из сепарационной части хлоратора, поступает на непрерывную ректификацию, в процессе которой хлорбензол и полихлориды отделяются, а бензол возвращается в цикл.
Выделяющийся хлороводород улавливается водой и в виде соляной кислоты используется в промышленности.
В условиях непрерывного процесса реакционная масса представляет собой газопарожидкостную эмульсию плотностью 200–300 кг/м3. Реактор работает в режиме полного вытеснения, благодаря чему достигается мольное соотношение хлорбензола к дихлорбензолу в реакционной массе, близкое к 40. Лучшего соотношения пока не удалось достичь.
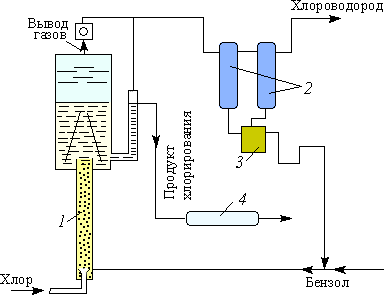
Рис. 5. Технологическая схема процесса непрерывного хлорирования бензола: 1 — колонна с катализатором (хлоратор); 2 — конденсаторы-холодильники; 3 — приемник обратного бензола; 4 — холодильник
Из полихлоридов, комбинируя кристаллизацию и вакуум-перегонку, выделяют о- и п-дихлорбензолы, 1,2,3- и 1,2,4-трихлорбензолы, а также 1,2,4,5-тетрахлорбензол, которые находят значительное по объему промышленное применение. В меньших масштабах выделяют и используют также пента- и гексахлорбензолы.
Для получения хлорбензола в промышленности применяется также парофазный каталитический метод — так называемый метод окислительного хлорирования. Реакцию осуществляют, пропуская пары бензола и хлороводород при 200–250 °С над катализатором, содержащим медь(II). В присутствии кислорода воздуха идет окисление хлороводорода и одновременное хлорирование бензола по суммарному уравнению (4.1.41).
С6Н6 + HCl + 0,5O2 > С6Н5Cl + Н2О (4.1.41)
Степень превращения бензола за проход — около 10 %, выход хлорбензола — 90 % от теоретического. Одно из преимуществ этого метода заключается в практическом отсутствии полихлоридов, а основной его недостаток — трудность подбора материалов для аппаратуры, которая должна работать в среде хлороводорода и воды при высокой температуре.
Гексахлорбензол (20) в промышленности получают хлорированием бензола при 300 °С (в присутствии катализатора (IrCl2 или RhCl3 на Al2O3)) или при 600 °С, а также окислительным хлорированием бензола смесью НСl с воздухом при 200–250 или 300–400 °С в зависимости от катализатора (соли меди, активированные Al или Fe).
Получение нитропроизводных бензола
Из нитропроизводных бензола, полученных прямым нитрованием бензола, практическое значение имеют нитробензол и м-динитробензол. Нитробензол (23) в основном используется для получения анилина, потребность в котором постоянно возрастает; производство нитробензола крупнотоннажное и осуществляется непрерывным методом. м-Динитробензол (24) используется в значительно меньших масштабах, поэтому производится преимущественно периодическим способом. Из м-динитробензола получают м-нитроанилин и м-фенилендиамин. В настоящее время потребность в м-фенилендиамине значительно возросла в связи с его применением для производства термостойких полимеров и волокон.
Нитробензол в промышленности получают нитрованием бензола нитрующей смесью, в которой количество нитрующего агента несколько меньше теоретического. Это необходимо для того, чтобы азотная кислота была использована полностью и в продукте нитрования не было примеси динитросоединений. Небольшое количество не вступившего в реакцию бензола легко отделяется при перегонке.
Непрерывное нитрование бензола проводится в нитраторе 5 — стальном аппарате, снабженном рубашкой, двумя внутренними змеевиками для охлаждения водой и двумя мешалками — турбинной и пропеллерной, посаженными на один вал. Верхняя мешалка (турбинная) служит для интенсивного смешения подаваемых на нее реагентов: бензола, кислотной нитросмеси и отработанной кислоты; нижняя мешалка (пропеллерная) работает как осевой насос. Бензол, нитрующая смесь и отработанная серная кислота вводятся в нитратор непрерывно через дозирующие устройства из напорных баков 2, 3, 4 соответственно. Подача бензола и нитросмеси автоматически регулируется по заданному соотношению, а количество отработанной кислоты — по температуре в реакторе, которая поддерживается на уровне 65–68 °С. Нитратор работает в режиме полного смешения. При установившемся процессе концентрация нитробензола в смеси постоянна и равна 5 %.
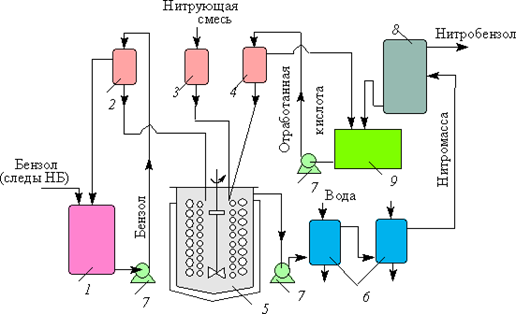
Рис. 6. Технологическая схема процесса непрерывного нитрования бензола: 1 — емкость для бензольного экстракта; 2, 3, 4 — напорные баки для бензола, кислотной смеси и отработанной кислоты; 5 — нитратор; 6 — теплообменники; 7 — насосы; 8 — сепаратор; 9 — сборник отработанной кислоты
Из нитратора реакционную массу подают с помощью насоса 7 в два расположенных последовательно спиральных теплообменника 6. В первом из них завершается реакция нитрования, во втором температура реакционной массы снижается до 25–30 °С. Охлажденную реакционную массу подают в сепаратор непрерывного действия 8, в котором происходит разделение нитробензола и отработанной кислоты. Так как разность плотностей нитробензола и отработанной кислоты велика, расслаивание происходит быстро (время пребывания реакционной массы в сепараторе 5–10 мин). Сырой нитробензол промывают водой, нейтрализуют аммиачной водой или раствором карбоната натрия; при этом легко удаляются минеральные кислоты. Нитрофенолы, образующиеся в качестве примеси, отмываются с трудом и при большом расходе воды. При нейтрализации аммиачной водой аммониевые соли нитрофенолов выпадают в осадок, который отделяют и утилизируют. В ряде случаев нитробензол без очистки передают на дальнейшую переработку — восстановление или сульфирование. Если нитробензол необходим в чистом виде, его очищают перегонкой в вакууме.
С каждой той товарного нитробензола из производственного цикла выводится 900–1000 кг 70–73% отработанной серной кислоты, содержащей 1,5–2,2 % нитробензола и 0,25–0,50 % азотной кислоты. Перед утилизацией этой кислоты из нее экстрагируют нитробензол. Экстракцию ведут бензолом, который при этом частично нитруется азотной кислотой, содержащейся в отработанной серной кислоте. В результате двухступенчатой экстракции полуторным по массе количеством бензола и последующего разделения слоев нитробензол практически полностью извлекается, а азотная кислота расходуется на нитрование бензола. Бензол со следами нитробензола собирают в емкость 1, а затем направляют в нитратор 5. Циркуляция нитробензола вызывает образование и накопление заметных количеств динитробензола. Наличие в нитробензоле даже 0,3 % динитробензола представляет опасность для процесса парофазного каталитического восстановления нитробензола до анилина, т. к. взрывоопасный динитробензол может накапливаться в испарителе. Отработанная серная кислота идет на регенерацию или используется для получения суперфосфата.
Получение нитробензола по непрерывному методу требует точного соблюдения технологического режима. Повышение температуры ускоряет процессы окисления. Сравнительно небольшой избыток азотной кислоты в нитраторе, появившийся в результате нарушения соотношения подаваемых реагентов, может привести к увеличению примеси динитробензола. Несоблюдение режима промывки и нейтрализации сырого нитробензола приводит к образованию стойких эмульсий и увеличению примесей нитрофенолов в готовом продукте и т. д. Лишь надежная автоматизация всего процесса может гарантировать высокую производительность синтеза и получение нитробензола высокого качества.
1,3-Динитробензол (м-динитробензол) получают в относительно небольшом количестве, поэтому производство его ведется периодическим способом в специальных стальных аппаратах (нитраторах) с большой поверхностью теплообмена в виде рубашек, змеевиков или полых цилиндров, в которые подается вода или холодильный рассол. Нитратор снабжается также быстроходной мешалкой, термопарой для непрерывной регистрации температуры и автоматическим устройством, закрывающим подачу нитрующего агента при прекращении размешивания массы или ее перегреве. Для получения 1,3-динитробензола используют нитрующую смесь, содержащую 33–34 % HNO3 и 66–67 % H2SO4. Азотную кислоту берут с небольшим избытком (на 1 моль бензола 2,05–2,10 моля HNO3). Нитрование начинают при 10–30 °С, а заканчивают при 90 °С. При этой же температуре производят отстаивание реакционной массы и отделение отработанной кислоты. Динитробензол промывают горячей водой, разбавленным раствором гидроксида натрия до нейтральной реакции и выделяют гранулированием в холодной воде. В полученном продукте содержится около 90 % мета-, 8–9 % орто- и 1–2 % пара-изомера. Для выделения м-динитробензола нитропродукт при 65–70 °С обрабатывают раствором сульфита натрия. При этом орто- и пара-изомеры превращаются в соли соответствующих нитробензолсульфокислот и переходят в раствор. м-Динитробензол в этих условиях не взаимодействует с сульфитом натрия.
м-Динитробензол можно выделить также с использованием кристаллизации. После отделения мета-изомера образовавшуюся эвтектическую смесь обрабатывают серной кислотой; при последующем охлаждении из нее можно выделить также о-динитробензол.
Получение сульфопроизводных бензола
При сульфировании бензола получают бензолсульфокислоту (25) и бензол-1,3-дисульфокислоту (27).
Производство бензолсульфокислоты крупнотоннажное, бензол-1,3-дисульфокислота производится в значительно меньших масштабах.
Бензолсульфокислота — основной продукт сульфирования бензола. Существует много промышленных способов получения бензолсульфокислоты, из них наиболее экономичным является сульфирование бензола «в парах». Технологическая схема сульфирования приведена на рис. 4.1.17.
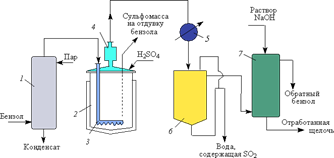
Рис. 7. Технологическая схема процесса сульфирования бензола «в парах»: 1 — испаритель; 2 — сульфуратор; 3 — кольцевой барботер; 4 — брызгоуловитель; 5 — холодильник-конденсатор; 6 — отстойник-сепаратор; 7 — нейтрализатор
Бензол из хранилища насосом подают во внутренние трубы испарителя 1 (типа «труба в трубе» или кожухотрубчатого). Во внешние трубы испарителя (в рубашку) подается греющий пар давлением 0,8 МПа. В испарителе бензол нагревается, испаряется, а пары его перегреваются до 150–160 °С (давление паров бензола на выходе из испарителя 0,05–0,08 МПа). Перегретые пары бензола, взятого в большом избытке (6–8-кратном), с помощью кольцевого барботера 3 поступают в сульфуратор 2, в котором барботируются через слой предварительно загруженной 94–96% серной кислоты (купоросное масло). Температура в сульфураторе поддерживается ~150 °С (обогревают через рубашку паром). Избыток бензола с парами воды через брызгоуловитель 4 поступает в холодильник-конденсатор 5, в котором смесь бензола и воды конденсируется и охлаждается. Охлажденная до 30–35 °С смесь поступает в отстойник-сепаратор 6, где расслаивается на составные части. Бензол после нейтрализации в колонне 7 возвращается на сульфирование.
Так как избыток бензола уносит из аппарата воду (как образовавшуюся при сульфировании, так и внесенную с купоросным маслом), концентрация серной кислоты в сульфураторе не снижается до значения π-сульфирования. Теоретически можно было бы использовать эквимольное количество серной кислоты, однако этому препятствует образование сульфонов. Чтобы их количество не превышало 1 %, сульфирование прекращают при содержании в реакционной массе 4–5 % непрореагировавшей серной кислоты.
По окончании сульфирования в сульфомассе остается 1,5–2 % бензола, который удаляют отдувкой воздухом, после чего сульфомасса, содержащая бензолсульфокислоту (88,5–91,5 %), бензол-1,3-дисульфокислоту (0,4–0,6 %), H2SO4 (3,0–3,5 %), дифенилсульфон (1,0 %) и бензол (0,2 %), поступает на нейтрализацию. Нейтрализация проводится непрерывным способом в стальном футерованном кислотоупорной плиткой аппарате с помощью водного раствора сульфита натрия, после чего реакционная масса поступает в колонну для удаления выделяющегося диоксида серы (его используют для выделения фенола).
После нейтрализации и удаления диоксида серы получается водный раствор, содержащий бензолсульфонат натрия (~50 %), сульфат натрия (~4 %), сульфоны (0,7%) и бензол-1,3-дисульфонат натрия (0,5 %). Этот раствор может быть направлен для щелочного плавления и получения фенола. Если необходимо, упариванием этого раствора можно получить сухой бензолсульфонат натрия.
При сульфировании бензола «в парах» расход серной кислоты снижается в 1,8–1,9 раза по сравнению с жидкофазным сульфированием. Недостатком сульфирования «в парах» является замедление сульфирования вследствие уменьшения содержания серной кислоты в реакционной массе.
Жидкофазное сульфирование бензола осуществляют действием двукратного количества моногидрата в периодически действующих сульфураторах при температуре ~60 °С в начале процесса и до 105 °С в конце процесса. Реакционную массу после разбавления водой частично нейтрализуют сульфитом натрия, а затем карбонатом кальция. Раствор бензолсульфоната натрия после фильтрации упаривают досуха.
Известны и другие методы сульфирования бензола. Один из непрерывных методов состоит в том, что жидкий бензол пропускают через серную кислоту (противотоком). Образующаяся бензолсульфокислота растворима в бензоле и вымывается затем из бензольного слоя водой.
Описано сульфирование бензола до бензолсульфокислоты слабым олеумом при 190–250 °С и давлении 1–3 МПа; образование сульфонов может быть подавлено добавлением бензолсульфоната натрия. В качестве сульфирующего агента для получения бензолсульфокислоты используют также 100%-й триоксид серы.
Промышленные методы сульфирования бензола были разработаны в основном для производства фенола: полученную бензолсульфокислоту действием NaOH и последующим подкислением превращали в фенол. В настоящее время эти методы потеряли свое прежнее значение в связи с развитием производства фенола из изопропилбензола.
Бензол-1,3-дисульфокислота производится в значительно меньших масштабах, поэтому получают ее в промышленности периодическим способом в специальных чугунных или стальных аппаратах (сульфураторах) со специфическим днищем и крышкой. Сульфуратор снабжен рубашкой для обогрева и охлаждения, мешалкой, термометром в гильзе, трубой для передавливания массы, в крышке имеются загрузочные отверстия. При прямом сульфировании бензола олеумом до бензол-1,3-дисульфокислоты получается значительное количество сульфонов, поэтому процесс обычно ведут в два этапа: вначале бензол сульфируют моногидратом при 50–60 °С, а затем 65% олеумом при той же температуре. Иногда применяется одностадийный способ получения бензол-1,3-дисульфокислоты, в этом случае сульфирование бензола 65% олеумом проводится в присутствии сульфата натрия, подавляющего образование дифенилсульфона. По окончании сульфирования сульфомассу передавливают в аппарат, содержащий раствор сульфата натрия, и нейтрализуют серную кислоту гидроксидом кальция (известковым молоком) или карбонатом кальция (мелом). После отделения сульфата кальция (гипса) и перевода кальциевой соли бензолдисульфокислоты в натриевую последнюю выделяют, выпаривая воду. Если бензол-1,3-дисульфокислота необходима для получения резорцина, после фильтрации раствор ее натриевой соли выпаривают до 60%-й концентрации и в этом виде используют для щелочного плавления.
Глава 2. ТРИНИТРОТОЛУЛ 2,4,6-ТРИНИТРОТОЛУОЛ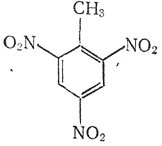
Μ = 227,23
Применяется в качестве вторичного взрывчатого вещества для снаряжения боеприпасов как в чистом виде, так и в виде смесей и сплавов с гексогеном, тетранитропентаэритритом, аммиачной селитрой (амматолы), динитронафталином и невзрывчатыми веществами — алюминием (алюмотол) и другими, а также в составе взрывчатых смесей для горнорудной промышленности.
Получается нитрованием толуола.
Физические свойства и состав. Желтоватые кристаллы, постепенно темнеющие на воздухе. Раств. в воде 0,021% (15°), в спирте 1,6% (22°). Технический тринитротолуол (т. плавл. 78—79°) может содержать другие изомеры тринитротолуола, динитротолуолы, динитробензол, тринитроксилол, тринитробензол, соединения фенольного характера, тетранитрометан.
Отравления возможны: 1) Путем вдыхания паров или пыли. В специальных цехах при плавке и на наиболее пыльных операциях общее поступление тринитротолуола через органы дыхания за 8-часовой рабочий день при умеренной физической работе колеблется от 75 до 100 мг, на большинстве операций оно не превышает 15—20 мг. 2) Через неповрежденную кожу. При нормальном выделении пота через кожу может поступить 200 мг тринитротолуола в сутки; при повышенном потоотделении 400—600 мг и более. 3) Путем заглатывания пыли. Общее количество тринитротолуола, которое может поступить в организм этим путем, не превышает 2,0—2,5 мг за сутки (Хоцянов).
Химически чистый тринитротолуол менее токсичен, чем технический препарат (Склянская, Пожариский). По данным Roberts, отравления амматолом связаны с высоким содержанием в нем нитрата аммония: последний, раздражая кожу, облегчает всасывание тринитротолуола По Koelsch, наибольшее значение имеет содержание тетранитрометана. В качестве весьма ядовитой примеси к тринитротолуолу называют также фенилтринитрометан.
Общий характер действия. Особенно характерны частые поражения печени вплоть до тяжелых токсических гепатитов, а также профессиональная катаракта. См. также Нитро- и аминосоединения ароматического ряда.
Токсическое действие. Животные. Острое отравление тринитротолуолом возникало у крыс при подкожном введении 0,9 г/кг, хроническое — при введении 0,2 г/кг. В обоих случаях нарушался обмен катехоламинов в головном мозге, сердце и надпочечниках (Кривицкая, Френкель; Мульменко). У крыс и кроликов в тех же условиях нарушался также углеводный обмен (Сонкин). У собак, отравляемых небольшими дозами тринитротолуола в течение 2 лет, выявлены расстройства функций желудка, печени и поджелудочной железы (Клейнер). Гистологически у погибших животных — жировая инфильтрация и вакуолизация в печени; дистрофические изменения в почках; воспаление слизистой желудка (Лаговиер). В сердце после 3 месяцев введения под кожу 0,2 г/кг — паренхиматозная дистрофия, набухание и гомогенизация отдельных мышечных волокон (Хижнякова и др.).
Человек. При острых отравлениях — синюшность губ, ногтей, головная боль, головокружение, тошнота. В более серьезных случаях — усиление синюшности, резкая слабость, тошнота, рвота, боли в правом подреберье. Тяжелые случаи протекают с резким цианозом, учащенным, поверхностным дыханием, тахикардией, сонливостью, потерей сознания, иногда с судорогами, поражениями печени разной степени. В крови метгемоглобин, тельца Гейнца, признаки гемолиза, ретикулоцитоз, базофильнозернистые эритроциты, пониженное содержание гемоглобина. Состояние общей кислородной недостаточности. Уменьшение артерио-венозной разницы, снижение содержания СО2 в артериальной и венозной крови. Газовый алкалоз, в тяжелых случаях — явления негазового ацидоза (Рашевская и др.). В более легких случаях отравление проявляется главным образом в желудочно-кишечных расстройствах, которые чаще носят функциональный характер. По утрам и после приема пищи — рвота, слюнотечение, отсутствие аппетита.
В производственных условиях в основном развивается хроническое отравление. В случае длительной работы при концентрациях тринитротолуола в воздухе на уровне ПДК или превышающих ее не более чем в 2—3 раза отмечаются вегетативно-сосудистая дисфункция (Айзенштадт) и сердечно-сосудистые расстройства с поражением сердечной мышцы, периферического кровообращения и гипотонией. Часты функциональные расстройства желудочно-кишечного тракта с нарушением желудочной секреции. Описано нарушение инкреторной функции поджелудочной железы, кортикостероидной деятельности надпочечников (Клейнер, Макотченко; Соболев; Хижнякова и др.; Панова; Каганов; Айзенштадт). Даже при наиболее легких формах интоксикации часто страдает печень (гепатит; нарушение антитоксической, белковообразовательной, липидной функций). При обследовании группы 100 и 200 человек гепатит выявлен у 11% (Каганов). В последние годы анемия определяется у 6—12% обследованных с хронической интоксикацией, ретикулоцитоз у 48% (Захарова, Каганов). У 26% из 104 взрывников при постоянном контакте с тринитротолуолом и концентрации последнего в воздухе в 2—2,5 раза выше ПДК в крови обнаружено более 4% метгемоглобина. Обнаружены также нарушения обмена серусодержащих веществ, по-видимому вследствие нарушения метаболизма цистина (снижение содержания цистина в ногтях). Дефицит витамина В6 выражался в повышенном выделении с мочой ксантуреновой кислоты и снижении экскреции 4-пиридоксиновой кислоты (Артамонова и др.).
Наиболее ранним, а иногда единственным признаком отравления тринитротолуолом является поражение хрусталика: обнаружено у 51 из 163 обследованных без других признаков интоксикации (Захарова, Манойлова). Частота поражения хрусталика повышается со стажем и со степенью контакта с тринитротолуолом. Так, при осмотре 221 взрывника помутнение хрусталика обнаружено почти у 80% (Пеньков), у половины из нескольких сот человек, работавших в контакте с тринитротолуолом (Захарова, Манойлова; Hassman, Juran), у 13% из 1000 взрывников железорудных шахт (при этом при стаже 4—5 лет частота профессиональной катаракты составляла 7,6%, а при большем — 35,6%). В среднем частота катаракты была 17% среди взрывников, а среди лаборантов, подсобников 4,7% (см. «Клиника, диагностика и профилактика тринитротолуоловых интоксикаций...»). Катаракта от тринитротолуола имеет особый характер: в начальной стадии образуется неясное кольцо помутнения по периферии хрусталика, постепенно расширяющееся и распространяющееся к центру, захватывая оптическую зону. Развитие катаракты довольно медленное, вначале острота зрения страдает мало. Есть указания, что поражаются и другие отделы глаза — стекловидное тело, сетчатка. Мнения о возможности обратного развития противоречивы (Пеньков; Захарова, Манойлова; Резников; Hassman, Juran).
У женщин — нарушение менструаций (Лаговиер).
Чувствительноетъ к отравлению неодинакова. Более чувствительны молодые люди, женщины, алкоголики, а также работающие натощак. Прием даже небольших доз алкоголя может выявить имеющееся в скрытой форме отравление. Возможно, гепатиты со смертельным исходом объясняются особой чувствительностью печени, например после перенесенных инфекций (тифы, малярия, сифилис) или отравлений (алкоголизм).
Действие на кожу. Тринитротолуол окрашивает кожу и волосы в желтый цвет. Вызывает дерматиты, особенно в первые месяцы работы, крапивницу. Поражаются лицо, шея, ступни и лодыжки, область половых органов. Описан гиперкератоз (Стоянов, Иванов).
При попадании на кожу — немедленно снять загрязненную одежду и смыть тринитротолуол струей воды. При проявлении гастроэнтерита — промывание желудка водой или физиологическим раствором, прохладный крепкий чай, боржом. Первые 1—2 дня воздержаться от приема пищи. Затем 2—3 дня жидкая пища, слизистые отвары, кисели. Вводить 5% раствор глюкозы капельной клизмой и внутривенно. Внутрь витамин U (0,05 г). При раздражении дыхательных путей — теплые влажные ингаляции и полоскание горла 2—3% раствором питьевой соды, боржомом или масляные ингаляции (вазелиновое или персиковое масло) с 1% ментолом. Закапывать в нос 0,5% раствор дикаина с адреналином или 2% раствор эфедрина. Внутрь — теплое молоко с боржомом или содой, слизистые отвары. При сильном кашле — кодеин, дионин. При поражении глаз — обильное орошение чистой водой или изотоническим раствором с последующей инстилляцией (при резком раздражении) вазелинового масла; закапывание 1—2 капель 1% раствора новокаина или 0,5% раствора дикаина с адреналином (1:1000). При остром конъюнктивите — закапывание в глаза адреналина (1:3000) или 3% эфедрина, альбуцида или 1% синтомициновой эмульсии. При острых дерматитах — в течение 1—3 дней холодные примочки, свинцовая вода, буровская жидкость, растворы резорцина, таннина и др. На ночь накладывать индифферентную пасту (паста Лассара, ланолин) или жир.
Индивидуальная защита. Меры предупреждения. Промышленный фильтрующий противогаз марки А, респираторы РУ-60М; РПГ-67; «Лепесток»; «Астра-2» и др. Тщательная защита кожи: перчатки, спецодежда из пыленепроницаемой ткани, сменяемая не реже раза в неделю, нательное белье. При применении тротила, аммонита для взрывных работ в шахтах — кожаные перчатки. Спецодежду чистить химическими способами, а не стирать (Шушковский). Контроль за остатками тринитротолуола на коже или одежде (при нанесении 1 % раствора КОН в спирте или мыла с 5—10% сульфита появляется красное окрашивание). Для очистки кожи и ее защиты рекомендуют также крем, содержащий 10% окиси цинка и 1—2% окиси железа на казеиновой основе (Schwarz; Rodier). Есть указания о нейтрализации тринитротолуола сульфитом калия (Sutton). См. еще у Хоцянова.
Герметизация и механизация всех процессов, где использование тринитротолуола необходимо, исключающие непосредственный контакт с ним. В шахтных разработках следует заменить тринитротолуол менее опасными соединениями или свести его содержание во взрывном материале до минимума. По данным Шушковского, 50% тринитротолуола при взрывных работах в железорудных шахтах может быть заменено иданитами (смесь селитры с соляровым маслом). Применение гранулированных взрывчатых средств; использование дистанционной зарядки шнуров; механизация доставки взрывчатых материалов; сохранение целостности оболочек, их герметичность, использование для оболочек непарафиновых материалов. Механизация и автоматизация взрывных работ. При использовании тринитротолуола на снаряжательных заводах см. у Хоцянова и др.
Предварительные и периодические осмотры раз в 6 месяцев при производстве и применении тринитротолуола с обязательным участием терапевта, окулиста и невропатолога, с анализом крови на содержание гемоглобина, эритроцитов, лейкоцитов, билирубина [37]. Осмотр глаз в проходящем свете при максимальном расширении зрачка. В число противопоказаний к работе с тринитротолуолом, помимо предусмотренных [37], рекомендуют включить заболевания поджелудочной железы и надпочечников. См. также «Клиника, диагностика и профилактика тринитротолуоловых интоксикаций...»
Специальный рацион лечебно-профилактического питания (№ 4) и дополнительно 150 мг витамина С в сутки. Рекомендуется высококалорийная, богатая белками диета с ограниченным содержанием жиров и углеводов, а также применение витаминов группы В, особенно витамина B6 (Сонкин; Goodwin; Мульменко).
Определение в воздухе основано на колориметрии окраски, возникающей в ацетоновом растворе со щелочью. Метод неспецифичен, другие полинитросоединения, кроме динитробензола, мешают определению. Чувствительность 1 мкг в анализируемом объеме [58]. Определение тринитротолуола, тетрила и динитронафталина разработано Нифонтовой.
Тринитротолуол - белые кристаллы, желтеющие на свету. Наиболее важное бризантное ВВ для снаряжения боеприпасов (в чистом виде или в смеси с гексогеном, октогеном, тетранитропентаэритритом) и для взрывных работ (в смеси с NH4NО3 и А1). К удару, трению, прострелу пулей, огню, искре, химическому воздействию не чувствителен. Прессованный и порошкообразный тротил чувствителен к детонации и надежно взрывается от стандартных капсюлей-детонаторов, запалов. Минимальный инициирующий заряд: 100 мг ГМТД или 260мг гремучей ртути. Плавленый и чешуированный тротил имеет пониженную чувствительность к детонации и требует промежуточного детонатора в виде некоторого количества прессованного тротила. Тпл. 80,85°С, Твсп. 310°С, теплота взрыва 4,2 МДж/кг. Бризантность: 19 мм, фугасность 285 см3, плотность отливки: 1,66 г/смЗ. Горение при атмосферном давлении: C7H5N3O6 ® 2NO + 3CO + H2O + 4C + 1,5H2 + 1,5N2 + 400 ккал/кг Горение при высоком давлении: C7H5N3O6 ® 6CO + C + 2,5H2 + 1,5N2 + 632 ккал/кг Детонация: C7H5N3O6 ® 1,5CO2 + CO + 2H2O + 4,5C + 0,5H2 + 1,5N2 + 1240 ккал/кг Не вступает в реакцию с твердыми материалами (металл, дерево, пластмассы, бетон, кирпич и т.п.), не растворяется водой, не гигроскопичен, не изменяет своих взрывчатых свойств при длительном нагреве, смачивании водой, и изменении агрегатного состояния (в расплавленном виде). Под длительном воздействии солнечного света темнеет и несколько повышает свою чувствительность (теоретически). При воздействии открытого пламени загорается и горит желтым, сильно коптящим пламенем. Горение в замкнутом пространстве большого количества может перерасти в детонацию (теоретически, на практике это не встречается). Продукты взрыва токсичны. Со щелочами и щелочными металлами образует соли – тротилаты, которые очень чувствительны. Тротил является продуктом воздействия смеси азотной и серной кислот на толуол. На выходе получается чешуированный тротил (отдельные мелкие чешуйки). Из чешуированного тротила механической обработкой можно получить порошкобразный, прессованный тротил, нагреванием плавленный тротил. Тротил нашел самое широкое применение из-за простоты и удобства его механической обработки (очень легко изготавливать заряды любого веса, заполнять любые полости, резать и сверлить), высокой химической стойкости и инертности, невосприимчивости к внешним воздействиям. А значит, он очень надежен и безопасен в применении. В то же время он обладает высокими взрывными характеристиками. В смеси с гексогеном, тетрилом, ТЭНом тротил понижает чувствительность последних, а в смеси с амиачно-селитренными ВВ тротил повышает их взрывчатые свойства, повышает химическую стойкость и снижает гигроскопичность. Тротил в России является основным ВВ для снаряжения снарядов, ракет, минометных мин, авиабомб, инженерных мин и фугасов. Получение тринитротолуола проходит в три стадии: Схема установки для нитрования толуола на всех стадиях: 1-ая стадия; (получение мононитротолуола): В трёхгорлую колбу, снабжённую мешалкой, капельной воронкой, термометром и помещённую на водяную баню и заливают 100 грамм толуола. (Следует полностью исключить соединения и стыки приборов резиновыми шлангами и пробками, так как резина сильно набухает в толуоле и теряет свою первоначальную форму). Пустив в ход мешалку, в реактор в течение 30 минут добавляют из капельной воронки 255 грамм нитрующей смеси состава: H2SO4 – 55%, HNO3 – 28%, H2O – 17%. (Количество моногидрата HNO3 взятого на нитрацию должно составлять 105% от теоретически необходимого). Во время процесса поддерживают температуру таким образом, чтобы в реакторе был постепенный подъем температуры от комнатной (в начале слива) до 38 – 40°С (к концу слива). По окончании слива нитрующей смеси нагревом водяной бани постепенно в течение 15 – 20 минут доводят температуру реакционной массы до 50°С. При этой температуре производиться выдержка в течение 30 минут. Слив нитрующей смеси в начале нитрации необходимо производить медленно, так как благодаря активности свежей смеси при этом имеют место температурные скачки. К концу же нитрации, когда нитрующая смесь в значительной степени разбавлена отработанной кислотой, слив можно вести быстрее. В период выдержки происходит окончательное донитовывание толуола. По окончании выдержки реакционную смесь сливают в делительную воронку, где происходит расслаивание верхнего слоя – мононитротолуола от нижнего слоя – отработанной кислоты. Продукт реакции - мононитротолуол, светло - жёлтая жидкость, температура плавления: 5°С. Выход мононитротолуола должен составлять не менее 95% от теоретически возможного.
2-ая стадия; (получение динитротолуола): В трёхгорлую колбу, снабжённую мешалкой, капельной воронкой и термометром заливают 220 грамм нитрующей смеси состава: H2SO4 – 67%, HNO3 – 23%, H2O – 10%. и помещают на водяную баню.(Количество моногидрата HNO3, взятого на нитрацию, должно составлять 200% от теоретически необходимого). Затем постепенно сливают при работающей мешалке 100 грамм мононитротолуола. Нагревом или охлаждением водяной бани регулируют температуру содержимого реактора, таким образом чтобы температура постепенно возрастала, начиная с комнатной и заканчивая 70 – 80°С. (Слив мононитротолуола продолжается от 30 минут до 1 часа). После слива всего мононитротолуола постепенным нагревом водяной бани повышают температуру реакционной смеси до 100°С и делают при этой температуре выдержку в течение 30 минут. Далее охлаждают содержимое реактора до 70 – 80°С и медленно прибавляют 100 мл воды. Скорость прилива воды регулируют таким образом, чтобы выделившийся динитротолуол был всё время в расплавленном состоянии (температура не ниже 70°С) и, чтобы с другой стороны не имел бы место слишком сильный подъём температуры (выше 90°С). Затем реакционная смесь сливается в предварительно подогретую делительную воронку, где в течение 5 – 10 минут происходит отделение слоя расплавленного динитротолуола (верхняя фаза) от отработанной кислоты. Слой динитротоуола сливают в фарфоровую чашку. 3-ая стадия; (получение тринитротолуола): В трехгорлую колбу емкостью 1 литр, снабженную термометром, капельной воронкой, механической мешалкой и помещённой на водяную баню, заливается 100 грамм расплавленного динитротолуола. Затем нагревают содержимое колбы до 75°С и при этой температуре начинают медленно сливать из капельной воронки нитрующую смесь в количестве 400 грамм состава: H2SO4 – 83%, HNO3 – 17%. Во время слива кислотной смеси содержимое колбы непрерывно перемешивается и нагревом или охлаждением водяной бани обеспечивается равномерный подъём температуры от 75° С до 85°С – к концу слива. Слив кислотной смеси должен продолжаться около 1 часа. Когда вся кислотная смесь слита, повышают температуру содержимого колбы до 110 – 115°С. (С этой целью сменяют водяную баню на заранее приготовленную нагретую до 90 – 100°С масляную баню). Период подъёма температуры должен продолжаться около 30 минут. После этого при температуре 110 – 115°С, выдерживают содержимое колбы в течение 1 часа, при непрерывной работе мешалки (3-ая стадия нитрации толуола сопровождается значительными окислительными процессами, проявляющимися в выделении окислов азота). При недостаточном соблюдении температурного режима нитрации возможны местные перегревы, которые могут повлечь за собой резкий температурный скачок. В этом случае необходимо прекратить слив нитрующей смеси и озаботиться о максимальном охлаждении реакционной массы. Не следует поднимать температуру выше 140°С). По истечении этого периода нитрация закончена. Содержимое колбы охлаждают до 80 – 85°С, и затем медленно при работающей мешалке приливают в реакционную колбу из капельной воронки около 100 мл воды. Время слива воды регулируют таким образом, чтобы температура содержимого колбы находилась в пределах 90 – 100°С. По окончании слива воды содержимое колбы переливается в предварительно подогретую делительную воронку ёмкостью в 1 литр, где в течение 3 – 5 минут происходит отделение слоя расплавленного тротила (верхний слой) от отработанной кислоты (нижний слой). Слой расплавленного тротила сливается в фарфоровую чашку. Выход сырого кислого тротила составляет 110 – 115 грамм. Температура затвердевания хорошо пронитрованного продукта не ниже 78°С. Отмывка кислого тротила от кислот: Полученный тротил содержит значительное количество кислых примесей, как минерального, так и органического характера. Для удаления последних применяют промывку тротила от кислот в расплавленном состоянии горячей водой. В автоклав ёмкостью 500 см3, снабжённый мешалкой, помещают 50 – 100 грамм кислого тротила и тройное по весу количество горячей воды. Расплавившийся тротил перемешивают с горячей водой в течение 5 – 10 минут. Затем останавливают мешалку и осторожно сливают сифоном кислую воду. Затем заливают в автоклав свежую порцию горячей воды и снова повторяют промывку по вышеописанному способу. Операцию промывки проводят несколько раз, пока промывные воды не покажут нейтральной реакции на лакмус. После этого тщательно отделяют сифоном последнюю промывную воду и расплавленный тротил сливают в тарированную фарфоровую чашку. После этого тротилу дают остыть в течение 1 часа до комнатной температуры, сливают находящуюся на поверхности воду и помещают в сушильный шкаф, в котором сушат при 110°С в течение 6-ти часов. После этого высушенный тротил остужают в эксикаторе над CaCl2 до комнатной температуры. Сульфитная очистка тротила: Сухой неочищенный тротил тонко измельчается в фарфоровой ступке. 50 грамм измельчённого тротила помешается в автоклав емкостью 500 см3, снабжённый механической мешалкой и термометром. Туда же заливается 100 мл свежеприготовленного водного раствора сульфида натрия, при этом раствор принимает красную окраску. Затем нагревом водяной бани доводят содержимое до 40° – 50°С, при этой температуре перемешивают в течение 1 часа, после чего переносят содержимое колбы на воронку Бюхнера, где тротил тщательно отжимают от промывных вод и промывают теплой (40° – 50°С) водой до полного обесцвечивания промывных вод. После этого очищенный тротил пересыпается в фарфоровую чашку, в которой сушиться при 110°С в течение 6-ти часов. Выход очищенного продукта 45 – 47 грамм. Перекристаллизация из спирта: В колбу, соединённую обратным холодильником и помещённую на водяную баню, помещают 50 грамм сырого тротила и 250 – 300 мл 96%-ного спирта. Нагревом водяной бани доводят спирт до кипения, при этом тротил растворяется в спирте. После этого полученный раствор (окрашенный в красный цвет) фильтруют, и дают остужаться до следующего дня. После этого выделившиеся кристаллы отфильтровывают на воронке Бюхнера и промывают небольшим количеством чистого спирта. Отжатые от спирта кристаллы сушат при 40 – 50°С. Выход кристаллического продукта 42 – 44 грамма.
Глава 3. ИЗВЛЕЧЕНИЕ СУЛЬФИТ НАТРИЯ ИЗ ОТХОДОВ ПРОЦЕССА ПРОИЗВОДСТВА ТРИНИТРОТОЛУОЛА
Сточные воды процесса производства тринитротолуола (ТНТ), окрашенные в красный цвет, содержат сульфит натрия, который может быть выделен из раствора. Согласно процессу, разработанному В.Р. Куком, эти сточные воды подвергают обработке для выделения соединений натрия без рецикла золы, образующейся при сжигании, и без формования из нее твердых гранул.
Смесь гидроксида алюминия и сточных вод, содержащих щелочной металл, пропускают чрез горячую печь, где происходит их сушка и образование гранул. При сгорании органических компонентов гранул протекает химическая реакция и получается гранулированная зола, содержащая алюминат щелочного металла. Алюминат натрия взаимодействует с отходящими газами, содержащими SOa, в результате чего образуется сульфит натрия и гидроксид алюминия, который возвращают в процесс.
Происходит окончательная сушка, а сжигание проводят в остальных секциях печи, где температура достигает 930 °С и выше.
Из нижней части печи выводят золу (550 кг/ч), которую охлаждают до 200 °С воздухом, подаваемым в холодильник противотоком. Золу затем измельчают и через уравнительный бункер направляют в резервуар для растворения, куда подается вода (1500 л/ч); в результате получают 25%-ный раствор алюмината натрия. Горячие газы, выходящие из печи, проходят через циклон, охлаждаются до 70 °С и подаются в абсорбер. При контактировании в абсорбере раствора алюмината натрия с охлажденными отходящими газами происходит осаждение гидроксида алюминия и образование сульфита натрия. Из абсорбера выводят суспензию гидроксида алюминия в растворе сульфита натрия при величине рН = 8,5. Ее промывают на ленточном фильтре; остаток от фильтрования удаляют и возвращают в первую секцию подовой печи для сушки. Фильтрат содержит ~385 кг сульфита натрия; после отстаивания его отводят в резервуар для хранения.
ЛИТЕРАТУРА
1. Лебедев Н.Н. Химия и технология основного органического и нефтехимического синтеза. М.: Химия, 1969. 670 с.
2. Адельсон С.В. и др. Технология нефтехимического синтеза. М.: Химия, 1985. 607 с.
3. Горелик М.В., Эффос Л.С. Основы химии и технологии ароматических соединений. М: Химия, 1992. 640 с.
4. Чичибабин А.Е. Основные начала органической химии. М.: Государственное научно-техническое изд-во химической литературы, 1963. т.1 545 с.
5. Грейш А.А., Демыгин С.С., Кустов Л.М. Нанесенные вольфрам-циркониевые и цеолитные катализаторы // Катализ в промышленности, 2002, № 4 с. 176. Чичибабин А.Е. Основные начала органической химии. М.: Государственное научно-техническое изд-во химической литературы, 1957. т.2 614 с.12
0 комментариев