ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ РФ
ФГОУ ВПО "Алтайский государственный университет"
Биологический факультет
Кафедра физиологии человека и животных
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА
(реферат)
Барнаул – 2006
Содержание
Введение. 3
1. Этиология болезни. 4
1.1 Патогенез. Механизмы генетической предрасположенности. 4
1.2 Биохимия и морфология развития болезни. 8
2. Клинические проявления. 13
Заключение. 15
Список литературы.. 16
Введение
К настоящему времени описан ряд нейродегенеративных заболеваний, для которых характерно постепенно развивающееся разрушение различных структур мозга, вызванное массовой гибелью нейрональных и / или глиальных клеток, что сопровождается существенным нарушением всех сторон деятельности ЦНС. Среди таких заболеваний большого внимания заслуживает болезнь Альцгеймера (БА).
Заболевание описано еще в 1906 г. гистологом Алоисом Альцгеймером и позднее названо его именем. По данным американских исследователей, в США БА встречается у 2,0-2,5% населения в возрасте до 70 лет, в более старших возрастных группах частота заболевания увеличивается примерно вдвое на каждые пять лет. В нашей стране также отмечается БА у большого числа пожилых людей. Так, по данным Центра психического здоровья РАМН до 4,5-5,0% населения г. Москвы в возрасте 60-65 лет страдают деменцией альйгеймеровского типа. Болезнь поражает лиц всех рас и этнических групп; среди больных чуть больше женщин, чем мужчин, хотя это может быть связано с большей продолжительностью жизни женщин [1].
1. Этиология болезни
Большинство случаев БА имеют мультифакториальную природу и являются спорадическими. В то же время многочисленные популяционные исследования показали, что 25-40% случаев БА могут быть семейными, т.е. в семье пробанда имеется, как минимум, еще один больной с этим заболеванием. Важная роль генетических факторов в развитии БА подтверждается высокой конкордантностью по болезни среди монозиготных близнецов. Анализ большого числа семей с БА позволил установить бимодальное распределение значений возраста дебюта симптомов, причем условной границей между ранними и поздними семейными случаями болезни принято считать возраст 58 лет.
В ранних случаях семейной БА заболевание обычно наследуется как аутосомно-доминантный признак, связанный с повреждением одного основного гена. Поздние же семейные случаи БА являются гетерогенными; чаще всего в этих случаях имеется полигенно обусловленная предрасположенность к БА, при которой накопление повторных случаев болезни среди родственников связано с действием комплекса генетических и средовых факторов, общих для членов данной семьи. По некоторым оценкам, наследственные моногенные формы составляют в целом около 5-10% случаев БА [2].
1.1 Патогенез. Механизмы генетической предрасположенностиНа сегодняшний день удалось установить, что БА вызывается мутациями в 4-х генах, расположенных в хромосомах 1, 14,19 и 21 (табл.1) [1]. Идентифицированы 3 гена, мутации, которых приводят к развитию наследственных (аутосомно-доминантых) форм БА с ранним началом симптомов. Один из них - ген белка-предшественника β-амилоида, локализованный на хромосоме 21q 21 и обозначаемый аббревиатурой АРР (от англ. Amyloid Precursor Protein). Ген состоит из 19 экзонов, причем аминокислотная последовательность β-амилоида кодируется частью экзонов 16 и 17; данная аминокислотная последовательность расположена в карбоксильной части белка АРР. В норме белок АРР подвергается протеолизу под воздействием α-, β - и γ-секретаз; два последних протеолитических пути приводят к высвобождению интактных молекул β-амилоида, что само по себе не сопровождается развитием болезни.
Все восемь известных патогенных точковых мутаций АРР расположены в 16 и 17 экзонах гена и ведут к нарушению β - и γ-секретазного процессинга белкового продукта АРР, результатом чего является гиперсекреция пептида β-амилоида или преимущественная секреция более длинных, склонных к быстрой фибриллярной агрегации форм β-амилоида. В обоих случаях высвобождаемый пептид приобретает амилоидогенные свойства – процесс, лежащий в основе формирования сенильных бляшек в паренхиме мозга. В целом, мутации в гене АРР представляют большую редкость: во всем мире они выявлены лишь в 20 семьях и, по приблизительным оценкам, обусловливают не более 5% всех случаев семейной БА с ранним началом симптомов.
Два других гена, обусловливающих основную часть случаев ранне-семейной БА и расположенные на хромосомах 14q24.3. и 1q31-42, были клонированы в 1995 году. Эти гены являются высокогомологичными и кодируют родственные мембранные белки – пресенилины (соответственно, пресенилин-1 (РS1) и пресенилин-2 (PS2)). В мозге пресенилины экспрессируются преимущественно в нейронах и локализованы в ЭПР тел нейронов и их дендритов. Предполагается, что одна из функций пресенилинов может быть связана с регуляцией внутриклеточного транспорта мембранных белков, в т. ч. белка-предшественника β-амилоида. Мутации в генах пресенилинов сопровождаются гиперпродукцией амилоидогенных форм пептида β-амилоида, формирующих сенильные бляшки. Этот феномен обусловлен, наиболее вероятно, активизацией γ-секретазного протеолиза АРР в условиях "задержки" данного белка в ЭПР.
Другой возможный механизм патогенного эффекта мутантных пресенилинов может заключаться в индуцировании апоптоза вследствие нарушенной регуляции кальциевого гомеостаза в ЭПР и активации свободнорадикальных реакций. В этом случае выявляемое нарушение процессинга АРР в клетках, экспрессирующих мутантные пресенилины, носит вторичный характер по отношению к реализуемому "апоптическому каскаду". В целом, чуть более половины всех семейных случаев БА с ранним началом обусловлены мутациями в генах пресенилинов; при этом основная часть случаев связана с пресенилином-1, тогда как повреждения гена пресенилина-2 встречаются весьма редко (лишь 3 описанных мутации). Следует подчеркнуть, что 70% всех известных мутаций в генах пресенилинов являются уникальными (т.е. каждая из них была выявлена лишь в какой-то одной семье).
Большинство мутаций в генах АРР и пресенилинов характеризуются полной пенетрантностью к концу 6-го десятилетия жизни и неизбежно приводят к манифестации болезни при условии достижения носителем мутации соответствующего возраста. Анализ клинико-генетических корреляций показал отсутствие каких-либо существенных различий между фенотипами отдельных молекулярных форм БА, за исключением возрастных рамок появления первых симптомов болезни. При повреждении гена АРР заболевание манифестирует в возрасте 39-67 лет, несколько более позднее начало болезни наблюдается у больных с мутациями в гене пресенилина-2(50-65 лет), тогда как в случае мутаций в гене пресенилина-1 носит наиболее агрессивный и ранний характер (начало болезни от 24 до 56 лет). Некоторые мутации в гене пресенилина-1 могут в единичных случаях вызывать развитие атипичного фенотипа БА, характеризующегося сочетанием ранней деменцией с нижним спастическим парапарезом.
В значительном числе семей с ранней БА мутации в генах АРР и пресенилинов были исключены, что свидетельствует о дальнейшей генетической гетерогенности ранней формы заболевания.
Классическим примером выраженной генетической ассоциации является значение аполипопротеина Е как важнейшего эндогенного фактора риска в развитии поздней формы БА. Аполипопротеин Е (апоЕ) представляет собой белок с молекулярной массой 34 кДа, кодируемый геном на хромосоме 19q13.2. АпоЕ играет ключевую роль в метаболизме липидов (особенно холестерина), способствуя их перераспределению между клетками различных органов. В 1993 году установлено, что апоЕ является одним из протеинов, специфически связывающихся с β-амилоидом.
Ген апоЕ имеет 3 основных аллеля (ε2, ε3 и ε4), отличающихся единичными нуклеотидными заменами и определяющих существование 3 изоформ белка апоЕ, причем в общей популяции аллель ε3 наиболее распространен. В серии исследований, проведенных в 1993-1996 гг., было установлено, что аллель ε4 гена апоЕ встречается достоверно чаще у больных с поздней формой БА – как семейной (50%), так и спорадической (40%). Более того, риск развития на протяжении жизни БА в зависимости от генотипа апоЕ является доза-зависимым: у гомозиготных носителей аллеля ε4 он является наивысшим и составляет около 90%, у гетерозиготных носителей ε4 он равен 47%, тогда как лишь 20% лиц, не имеющих аллеля ε4, заболевают БА в пожилом возрасте. Доза "неблагоприятного" аллеля ε4 напрямую коррелирует также с интенсивностью формирования амилоидных бляшек в мозге больных с БА [2].
Таблица 1. Гены, связанные с болезнью Альцгеймера [1]
| Гены | Проявление болезни*, тип | Белок – продукт гена | Локализация гена |
| AD 1 | Раннее, наследуемый | АРР | 21q.21.2 |
| AD2 | Позднее, наследуемый\спорадический | АпоЕ | 19q.13.2 |
| AD3 | Раннее, наследуемый | Пресенилин-1 | 14q.24.3 |
| AD4 | Раннее, наследуемый | Пресенилин-2 | 1q.24.3 |
*Раннее проявление болезни – до 65, позднее – после 65 лет.
1.2 Биохимия и морфология развития болезниПри исследовании мозга умерших пациентов выявляется атрофия, особенно выраженная в ассоциативных зонах неокортекса, гиппокаипальных и парагиппокаипальных структурах наряду с заметным расширением латеральных желудочков. Наиболее значимым, "маркерным" признаком БА считается наличие многочисленных экстраклеточных амилоидных отложений (сенильные бляшки), располагающихся рядом с дегенерирующими аксонами и дендритами. Более всего сенильных бляшек встречается в коре и лимбических структурах, кроме того, амилоидные отложения наблюдаются в стенках кровеносных сосудов мозга – кортикальных и менингиальных артериях, артериолах, капиллярах и (в меньшей степени) в венах. Амилоидные отложения преимущественно локализуются на аблюминальной мембране этих сосудов. Число поврежденных амилоидными скоплениями сосудов может очень сильно варьировать в разных случаях БА при одинаковой "плотности" сенильных бляшек. Следует отметить, что подобные амилоидные отложения в небольшом количестве и с ограниченным распределением в лимбических структурах встречаются и в мозге пожилых людей, не страдающих БА.
В мозге большинства пациентов, умерших от БА, кроме сенильных бляшек обнаружены интранейрональные цитоплазматические нитчатые структуры – нейрофибриллярные сплетения. Чаще всего они присутствуют в телах тех нейронов, дегенерировавшие аксоны которых находятся в области сенильных бляшек.
Многочисленные нейрофибриллярные сплетения встречаются в нейронах ассоциативной и лимбической областей коры, а также в нейронах субкортикальных ядер. В то же время, подобные сплетения очень редко встречаются в других структурах мозга, которые минимально затронуты при БА, например, в мозжечке [1].
Амилоидные бляшки и нейрофибриллярные клубки – характерные, но не специфические признаки БА. Подобные изменения могут быть обнаружены у здоровых людей в процессе старения и при различных других нейродегенеративных болезнях [7].
Благодаря достижениям молекулярных биологов, генетиков, нейрохимиков за последнее десятилетие получен ряд принципиальных данных о биохимических механизмах, связанных с развитием БА. Проведен детальный анализ компонентов амилоидных отложений (сенильных бляшек), столь характерных для этого заболевания (табл.2).
Таблица 2. Химический состав сенильных бляшек при болезни Альцгеймера [1]
| Группы соединений | Вещества, обнаруженные в сенильных бляшках |
| Белки | Β-амилоидный белок (β-А) Протеогликаны: гепарин-сульфат, кератин-сульфат, дерматан-сульфат. Аполипопротеины: АпоЕ, АпоJ |
| Ферменты | Кислые протеазы(катепсины B,D) Ферменты метаболизма глюкозы: глюкозидаза, гексоамидаза Арилсульфатаза Кислая фосфотаза Холинэстераза Комплемент: C1q, С4, С5 |
| Ингибиторы протеаз | Ингибитор цистеиновых протеаз Ингибиторы сериновых протеаз: антихимотрипсин, антитрипсин, антитромбин |
| Вещества, содержащиеся в малых количествах | Продукты гликирирования Ионы металлов: Al2+, Zn2+ |
Основным компонентом, входящим в состав сенильных бляшек, является β-амилоидный белок, на его долю приходится до 25% сухого веса бляшек.
Присутствие в сенильных бляшках протеогликанов и аполипопротеинов интересно тем, что in vitro установлена их способность существенно ускорять фибриллогенез синтетического β-амилоида.
Обнаружение в сенильных бляшках ионов алюминия послужило основанием для предположения о токсическом действии этого элемента как причине БА. Однако более тщательные исследования, выполненные в последние годы, показали, что проникновение алюминия в мозг и связывание его с нейронами – явление вторичное, вызванное нарушением защитных функций гематоэнцефалического барьера.
Картину патологических изменений, связанных с резким возрастанием внутриклеточной концентрации ионов кальция при БА, дополняют нарушения, вызванные активацией ионами Са2+ калпаина – протеазы, основным субстратом которой служат нейрофибриллярные белки (тубулин, спектрин и др.) заметное повышение активности калпаина – характерный признак БА; при этом происходит разрушение цитоскелета нейронов и формирование нейрофибриллярных сплетений и тяжей [1].
В настоящее время выявлено несколько возможных биохимических механизмов развития данного заболевания, среди них: способность агрегированного β-А усиливать свободнорадикальные процессы мозге, способность его инициировать процессы апоптоза, повышение эксайтотоксичности возбуждающих аминокислот β-амилоидом, опосредованное накоплением β-А резкое нарушение гомеостаза Са2+ в нейронах и др. (сформулировано автором, цитир. по [1]).
Многие специалисты считают, что в патогенезе БА, как и ряда других нейродегенеративных заболеваний, участвуют в той или иной мере все описанные биохимические механизмы. Относительная роль каждого из них определяется индивидуальными особенностями организма и стадией патологического процесса.
Глубокие дегенеративные повреждения многих структур мозга при БА сопровождаются нарушением функционирования почти всех нейромедиаторных систем. При сенильной деменции гораздо сильнее (по сравнению с др. нейродегенеративными заболеваниями) выражено поражение холинергической системы. Установлено, что в мозге больных БА существенно замедлен выброс АХ из везикул в синаптическую щель, а также процесс обратного захвата холина [1].
Последовательность молекулярных событий, приводящих к развитию болезни Альцгеймера:
Миссенс-мутации в генах АРР, PS1,PS2
↓
Измененный протеолиз АРР
↓
Увеличенное образование β-А 42и\или общего β-А
↓
Прогрессирующее накопление нерастворимых агрегатов β-А 42 в межклеточном пространстве мозга
↓
Отложение агрегированного β-А 42 в виде диффузных бляшек
(в соединении с протеогликанами и др. амилоид-активизирующими субстратами)
↓
Агрегация β-А 42 в диффузные бляшки β-А 42
Накопление определенных белков, ассоциированных с бляшками
↓
"Воспалительный ответ":
•активация микроглии и высвобождение цитокинов
•астроцитоз и выброс белков
↓
Прогрессирующее разрушение нейритов
внутри амилоидных бляшек и в нейропиле
↓
Нарушение метаболического и ионного гомеостаза
в нейронах; окислительные повреждения
↓
Измененная киназная\фосфотазная
активность →гиперфосфорилирование τ →образование PHF
↓
Распространяющаяся нерональная \нейритная дисфункция и
гибель клеток гиппокампа и коры мозга с
прогрессирующим дефицитом нейротрансмиттеров
↓
деменция
Определенные успехи сделаны при поисках веществ, замедляющих агрегацию секретированного β-А в фибриллярную цитотоксическую форму. Поиски подобных веществ весьма перспективны, т.к их взаимодействие с β-А-ансамблями в экстраклеточном пространстве мозга поможет избежать вмешательства в метаболизм и функции растворимого фрагмента АРР [1].
2. Клинические проявления
Заболевание характеризуется прогрессирующим ослабоумливающим процессом, центральное место, в развитии которого занимают нарушения памяти, являющиеся наиболее ранним и типичным проявлением болезни. Спустя несколько лет от начала болезни закономерно присоединяются расстройства праксиса, речи, счета, письма, ориентировки и узнавания, у больных могут отмечаться острые психотические эпизоды, эпилептические припадки, разнообразные экстрапирамидные симптомы. В конечном счете, развивается глубокая тотальная деменция с распадом личности, тотальная афазия, общее физическое истощение. Средняя продолжительность заболевания составляет около 10 лет [2,3].
Наблюдается ослабление, прежде всего краткосрочной памяти, вплоть до потери способности ориентироваться в простейших бытовых ситуациях; расстройства эмоциональной сферы, когнитивных и двигательных функций. БА, постепенно прогрессируя, превращает еще достаточно крепких физически пожилых людей в беспомощных инвалидов, неспособных обслуживать себя и требующих постоянной опеки окружающих [1].
Современное состояние проблемы:
Клонирован и секвенирован новый ген человека, названный хьюмаином (humain, HN). Этот ген кодирует белок, предупреждающий гибель нейронов при мутациях в генах, вызывающих семейную БА – генах пресенилинов 1 и 2, гене АРР. Кроме того, хьюмаин влияет на гибель нейронов, вызываемую экспрессией пептида амилоида β-А [9].
В патогенезе БА могут играть роль провоспалительные цитокины и один из генов предрасположенности к развитию этого заболевания картирован на хромосоме 6 в области локализации гена одного из таких цитокинов, фактора некроза опухолей – TNF… [8].
У родственников пациентов с БА наблюдались признаки дисфункции глубоких структур мозга. Изменения более выражены у родственников пациентов с семейной формой БА [10].
Одним из факторов риска является низкий образовательный ценз (в 1.8. раза увеличивается). При ежедневном выпивании 3 стаканов вина риск возникновения БА в 2 раза меньше. К факторам риска относятся: высокая концентрация холестерина и липопротеинов высокой плотности, повышенная агрегационная способность тромбоцитов, несбалансированный режим питания. Профилактическое действие вина объясняется наличием в нем антиоксидантных полифенолов. На вероятность развития БА влияет степень умственной и физической активности [6].
Установлено, что в мозге пожилых людей и больных с БА в высокой концентрации содержится вирус простого герпеса 1-го типа (ВПГ-1). Отмечено, что ВПГ-1 представляет большой риск развития БА у лиц с носительством аллеля гена аполипопротеина Е. С помощью ПЦР проверяли содержание в мозге пациентов с БА других вирусов семейства герпеса: вируса герпеса 6-го типа, ВПГ-2 и ЦМВ. Отмечено. Что в мозге пациентов с БА в больших количествах содержится также вирус герпеса 6-го типа [11].
Диагностика ранних стадий БА остается сложной клинической задачей, но развитие радиоизотопных методов исследования головного мозга, особенно ПЭКТ, позволяет надеяться на скорое решение этой задачи. Проведенные исследования показали снижение церебрального кровотока и метаболизма глюкозы в височно-теменных областях у больных с ранними стадиями БА. Показано, что ПЭКТ выявляет БА с точностью >90% на 2.5г. раньше, чем это позволяют клинические диагностические методы, такие как ЭЭГ и др. [4].
Обсуждаются перспективы экспериментального поиска путей борьбы с БА и со связанными с нею нейродегенеративными расстройствами. Подчеркнута роль генетических, клеточно-биологических генно-инженерных исследований. Рассматривается и потенциал антиамилоидных вакцин, ингибиторов гамма-секретазы, блокаторов агрегации β-А, медевыводящих хелатов [5].
Заключение
В клинической медицине болезнь Альцгеймера является первым примером распространенного заболевания, для которого установлен ведущий генетический фактор предрасположенности. В связи с этим болезнь Альцгеймера может рассматриваться как своеобразная модель для разработки методологических аспектов ДНК-тестирования при мультифакториальных болезнях человека.
Прямая ДНК-диагностика болезни Альцгеймера представляет собой непростую задачу, что связано с генетической гетерогенностью, сравнительно большими размерами изучаемых генов и отсутствием в них мажорных мутаций. Опыт такой диагностики в мире имеется лишь в сравнительно небольшом числе хорошо оснащенных лабораторий, специализирующихся на молекулярно-генетическом анализе данного заболевания [2].
Несмотря на достаточно хорошо изученные генетические и биохимические механизмы развития болезни, до сих пор не найдены эффективные меры борьбы и предотвращения возникновения данной патологии, которые реально можно было бы применить на практике. Я думаю, это вопрос недалекого будущего, судя по интенсивности, с которой идут поиски в данном направлении.
Список литературы
1. Ещенко Н.Д. Биохимия психических и нервных болезней. – СПб.: Изд-во С-Петерб. Ун-та, 2004 – 200 с.
2. Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК-диагностика и медико-генетическое консультирование в неврологии. – М.: Мед. инф. аг-во, 2002 – 591 с.
3. Наследственные болезни нервной системы: рук-во для врачей /Под ред. Ю.Е. Вельтищева, П.А. Темина – М.: Медицина, 1998 – 496 с.
4. Радиоизотопное изображение головного мозга и диагностика болезни Альцгеймера/ Li Jian-Nan, Shang Yu-Kun // Di-er junyi daxue xuebao = Acad. J. Second Mil. Med. Univ. – 2003 – 24, №4 – c.447-450 (РЖ, Биология, Физиология человека и животных, Нейрофизиология, 2003 - №3).
5. Alzheimers disease and related dementias: The road to intervention // Exp. Gerontol – 2000 – 35, № 4, с.433-437 (РЖ, Биология, Генетика и цитология, Генетика неврологических заболеваний, 2003, №7).
6. Alzheimer: L’etude Paquid/ Letenneur L. // Biofutur. 2001. – Прил. Oct. – с.16 (РЖ, Биология, Физиология человека и животных, Общие и теоретические проблемы нормальной и патологической физиологии, 2002, №12).
7. А. rescue factor abolishing neuronal cell death bya wide spectrum of familial Alzheimers disease genes and Aβ /Hashimoto Yuichi, Niikura Takako… // Proc. Nat. Acad. Sci. USA – 2001 – 98, №11 – c.6336-6341/ (РЖ, Биология, Генетика и цитология, Генетика неврологических заболеваний, 2003, №12).
8. Association of a. haplotipe for tumor necrosis factor in siblings with late-onset Alzheimer disease. The NIMH Alzheimer disease genetics initiative/ Collins Julianne S.,Perry Rodney T. u. a. // Amer. J. Med. Genet. – 2000 – 96, №6 – с.823-830 (РЖ, Биология, Физиология человека и животных, Общие и теоретические проблемы нормальной и патологической физиологии, 2002, №12).
9. Atiologie und Pathogenese der Alzheimer – Demenz / Kratsch T., Peters J., Frölich L. // Wien. med. Wochenschr – 2002 – 152, № 3-4, с.72-76. (РЖ, Биология, Физиология человека и животных, Нейрофизиология, 2004 - №4).
10. EEG alteration in the relatives of patients with Alzheimers disease: Abstr.8 th world Congress on PsychiatriCongress on Psychiatric Genetics, Versailles/ Ponomareva N., Fokin V. – 2000 – 96,№4 – с.521 (РЖ, Биология, Генетика и цитология, Генетика неврологических заболеваний, 2003, №12).
11. Herpesviruses in brainand Alzheimers disease / Lin Woan-Ru, Wozhiak Matthew A.,Cooper Robert J., Wilcock Gordon K. // J. Pthol. -2002 – 197, №3 – с.395-402 (РЖ, Биология, Физиология человека и животных, Нейрофизиология, 2003 - №3).
Похожие работы
... которые по мере утяжеления деменции нарастают. Однако были установлены и определенные различия между группами больных с собственно болезнью Альцгеймера и сенильной деменцией альцгеймеровского типа. Эти различия определяются разной представленностью в структуре синдрома дефектов операциональных и различных регуляторных факторов, а также разной последовательностью вовлечения в патологический процесс ...
... полугода, подтверждается отсутствием побочных явлений и осложнений терапии. Помимо выраженного холинергического дефицита, который представляет собой наиболее раннее и выраженное проявление болезни Альцгеймера, установлены также недостаточность других нейротрансмиттерных систем, в частности,серотонинергической, глутаматергической, а также нарушения активности моноаминоксидазы (МАО) типа В. На ...
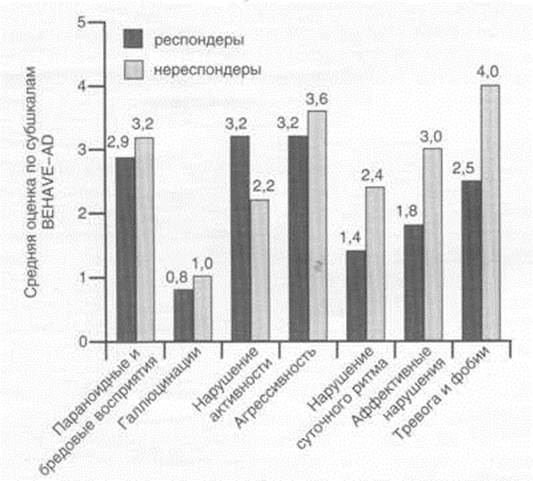
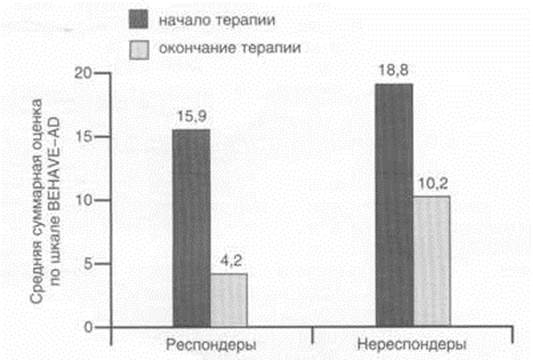
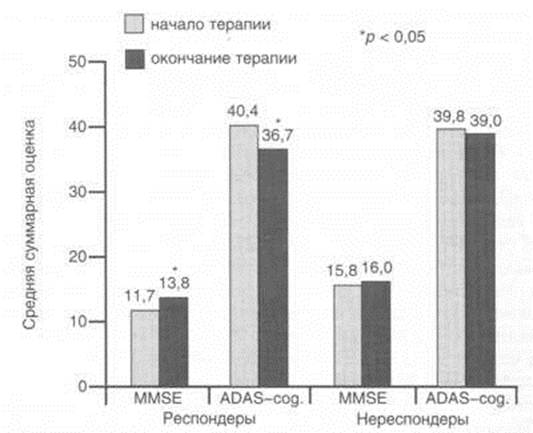
... исследования эффективности и безопасности применения кветиапина (сероквеля) для лечения психотических и поведенческих расстройств у больных БА. Исследование проводили в Научно-методическом центре по изучению болезни Альцгеймера и ассоциированных с ней расстройств НЦПЗ РАМН. Клиническое исследование выполняли как простое открытое на невыборочной группе пациентов с различными клиническими ...
... код внешних причин (класс XX). G71.2 Врожденные миопатии Врожденная мышечная дистрофия: · БДУ · со специфическими морфологическими поражениями мышечного волокна Болезнь: · центрального ядра · миниядерная · мультиядерная Диспропорция типов волокон Миопатия: · миотубулярная (центроядерная) · немалинная [болезнь немалинного тела] G71.3 ...
0 комментариев