Министерство образования Российской Федерации
Пензенский Государственный Университет
Медицинский Институт
Кафедра Терапии
Реферат
на тему:
"Дифференциальная диагностика нефротического синдрома при редких заболеваниях"Пенза
2010
План
1. Периодическая болезнь
2. Другие редкие причины нефротического синдрома
3. Патологические состояния, при которых нефротический синдром не встречается
Литература
1. Периодическая болезнь
а) Семейная Медитерранская лихорадка (периодическая болезнь)
Является наследственным заболеванием с наследованием по аутосомно-рецессивному типу. В развитии периодической болезни предполагается роль гена МЕFV, расположенного в коротком плече 16-й хромосомы. Известны и другие типы наследования этой болезни. Болеют в основном армяне, евреи и арабы, а также греки, кубинцы, итальянцы и бельгийцы. Наши наблюдения (18 случаев) связаны в основном с заболеванием армян, проживающих в Ростовской области в Мясниковском районе (село Чалтырь), а также Ростове-на-Дону. Наблюдали два случая заболевания турков, проживающих в общинах на территории области.
Первые клинические признаки болезни обычно появляются в раннем детском возрасте (3-6 лет) и представлены пароксизмально возникающей лихорадкой, диффузными болями в области живота, миалгиями, реже — артралгиями. Иногда возникают диффузные головные боли или боли по типу мигрени, тошнота, сухой кашель. Серозиты также носят пароксизмальный характер (асептический перитонит, плеврит, артрит). Поражение почек наблюдается уже в возрасте 5-25 лет. Появляется протеинурия, которая в последующем нарастает с переходом в нефротический синдром. Морфологически патологический процесс характеризуется развитием АА-амилоидоза. Поражение почек, как правило, демонстрирует рефрактерность к проводимой терапии.
У 90% пациентов первая атака развивается в возрасте до 20 лет. Симптомы болезни во время атаки персистируют непродолжительное время, в среднем от 6 до 96 часов. У 95% пациентов боль в животе продолжается 1-2 дня и может быть интенсивной с явлениями асептического перитонита, или слабовыраженной. Моноартрит с поражением коленного, голеностопного суставов, запястья наблюдается у 75% пациентов. Торакалгия с односторонним плевритом описана у 30% больных, перикардит — менее чем в 1% случаев.
б) Гипериммуноглобулинемия D с периодическим лихорадочным синдромом
Болезнь наследуется по аутосомно-рецессивному типу. Наблюдается мутация гена МVК. Первые случаи зарегистрированы в Нидерландах. У больных с гипер-IgD-синдромом развиваются периодические лихорадочные атаки. Дебют болезни наблюдается уже на первом году жизни. Атака проявляется ознобом с острым повышением температуры тела и продолжается в течение 4-6 дней. Может быть спровоцирована вакцинацией, небольшой травмой, хирургическим вмешательством или стрессом. Наблюдаются шейная лимфаденопатия, боли в животе с тошнотой, поносом. Выявляется гепатоспленомегалия, беспокоит головная боль, артралгии, развиваются артриты крупных суставов, эритематозно-папулезные или петехиальные высыпания. Интервалы между атаками длятся в среднем 4-6 недель. В крови регистрируется повышенное количество IgD (более 100 МЕ/мл). У 80% пациентов помимо повышения концентрации IgD повышается уровень IgА. При данном заболевании развитие амилоидоза почек характеризуется появлением протеинурии, которая постепенно нарастает, приводя к формированию нефротического синдрома.
в) ФНО-Р-ассоциированная периодическая болезнь
ФНО-Р-ассоциированная периодическая болезнь была описана как вариант болезни Стилла. Она наследуется по аутосомно-доминантному типу и связана с мутацией гена ТNFRSF1А (ген ФНО-Р массой 55 кД). Встречается в Ирландии и Шотландии. Известна также под названием семейная Хиберниальная лихорадка. Атака может продолжаться от 1-2 дней до нескольких недель. Заболевание развивается в детском и подростковом возрасте и включает в себя абдоминальные боли, плеврит, периорбитальный отек, миалгию, артралгию, боль в мошонке. Кожные проявления в виде мигрирующих эритематозных пятен, петехий и уртикарий встречаются у 3/4 больных. В крови наблюдается лейкоцитоз и рост концентрации острофазных белков, который проходит с купированием атаки. В 10% случаев развивается амилоидоз, в этиологии которого предполагается мутация гена цистеин-редуктазы.
2. Другие редкие причины нефротического синдрома
Семейный холодовой аутовоспалительный синдром
Относится к числу так называемых криопиринопатий. Наследуется по аутосомно-доминантному типу. Наблюдаются мутации генов СIАS1/NАLР3/РYРАF1, локализующихся в 1-й хромосоме. Проявляется лихорадкой, артралгиями и болями в животе, наступающими после легкого переохлаждения, а также уртикарной сыпью. При гистологическом исследовании волдырь инфильтрирован лимфоцитами, нейтрофилами и небольшим количеством тучных клеток, что демонстрирует его отличие от обычной аллергической уртикарной сыпи.
Синдром Muckle-Wells
Наследуется по аутосомно-доминантному типу. Мутации касаются генов СIАS1/NАLР3/РYРАF1. Клиническая картина характеризуется появлением уртикарной сыпи (волдырей), прогрессирующей глухотой и поражением почек и других паренхиматозных органов (амилоидоз). Амилоидоз почек сопровождается развитием нефротического синдрома с быстрым прогрессированием ХПН. Нередко наблюдается лихорадка, помимо глухоты описаны случаи глаукомы, косоглазия, артралгий. Тип наследования как доминантный, так и рецессивный. Уртикарная сыпь имеет сходную гистологическую характеристику с таковой при семейном холодовом аутовоспалительном синдроме.
При проведении дифференциальной диагностики следует обратить внимание на лихорадку, болевой синдром в животе и суставах, не характерные для нефротического синдрома, обусловленного хроническим или быстропрогрессирующим гломерулонефритом.
Нефротический синдром, ассоциированный с WT1-мутацией
При диффузном мезангиальном склерозе появляется протеинурия, ХПН развивается в течение первого года жизни. Морфологическая картина характеризуется гипертрофией подоцитов, дилатацией канальцев, тубулоинтерстициальным фиброзом. Диффузный мезангиальный склероз, ассоциирующийся с мужским псевдогермафродитизмом и опухолью Вильямса, именуется синдромом Denis-Drash. Ген, мутация которого приводит к развитию опухоли Вильямса, WT1 локализуется в 13-м локусе 11-й хромосомы. WT1-ген отвечает за эмбриональное развитие почек и гонад. Мутация WT1-гена обнаруживается также у детей, страдающих несемейным стероидрезистентным нефротическим синдромом. При этом наблюдаются врожденные аномалии (проксимальная гипоспадия, диафрагмальная грыжа, односторонняя эктопия яичка). Совокупность перечисленных признаков именуется синдромом Frasier. Синдром Frasier характеризуется полным ХY-гонадным дисгенезом в сочетании с нефротическим синдромом. Прогрессирование ХПН при этом синдроме более медленное, чем при синдроме Denis-Drash.
Синдром Pierson
Развивается при мутации гена 21 -го локуса 3-й хромосомы, кодирующего синтез р2-цепи ламинина. Характеризуется аутосомно-рецессивным типом наследования. Дебютирует нефротическим синдромом вскоре после рождения. Развиваются диффузный мезангиальный склероз и микрокория (резкое уменьшение размеров зрачков). Характеризуется крайне тяжелым течением и скорым летальным исходом на протяжении первых двух месяцев жизни.
Синдром Nail-раtеllа (синдром поражения ногтей и надколенника, синдром Турнера-Кизера).
Характеризуется аутосомно-доминантным типом наследования. Встречается с частотой 1:50 000 живорожденных. Развивается при мутации гена 1МХ1В, расположенного в 34-м локусе 9-й хромосомы и регулирующего экспрессию белков подоцитов. Характеризуется нефротическим синдромом в сочетании с симметричными аномалиями ногтей, скелета и глаз. В раннем детском возрасте обращает на себя внимание мягкость, тонкость и изогнутость ногтей или их полное отсутствие. Позже становится заметным уменьшение размеров коленных чашечек, что сопровождается затруднением ходьбы. Нередко это приводит к варусной деформации и артрозу коленных суставов. Может также развиваться сколиоз. Поражение почек наблюдается в 30-40% случаев и клинически проявляется протеинурией с развитием нефротического синдрома, реже — низкой протеинурией в сочетании с эритроцитурией или лейкоцитурией. Иногда нефротический синдром бывает врожденным.
Синдром Альпорта
В клинической картине синдрома Альпорта редко встречается нефротический синдром. Однако он может быть одним из проявлений болезни. При этом нередко сочетается с эритроцитурией. Также важно иметь в виду наличие и других проявлений в виде глухоты, реже — поражения органа зрения. Наследуется синдром Альпорта доминантно, чаще сцепленно с Х-хромосомой. Болезнь более тяжело протекает у мальчиков, чаще сопровождаясь развитием ХПН. Следует отметить, что нефротический вариант течения синдрома Альпорта является менее благоприятным, чем классическая гематурическая форма.
Синдром Галовэй-Мовата
Болезнь наследуется по аутосомно-рецессивному типу и проявляется врожденной микроцефалией, гормонорезистентным нефротическим синдромом, отставанием психомоторного развития. Реже также регистрируются диафрагмальная грыжа, катаракта, аномалии лицевого черепа. При гистологическом анализе могут наблюдаться разнообразные воспалительные изменения клубочков в виде фокально-сегментарного гломерулосклероза, липоидного нефроза, диффузного мезангиального склероза и др. Быстрое прогрессирование ХПН приводит к летальному исходу в раннем детском возрасте.
Синдром Лоу (окулоцереброренальный синдром)
Для синдрома Лоу характерен Х-сцепленный рецессивный тип наследования (мутация гена в 25-м локусе), значительно реже предполагаются другие типы наследования. В связи с этим страдают болезнью в основном мальчики, однако описаны случаи заболевания девочек. Синдром Лоу проявляется тяжелой умственной отсталостью, врожденной катарактой или глаукомой, гипотонией и почечными аномалиями в виде синдрома Фанкони с последующим постепенным развитием ХПН. Протеинурия быстро нарастает с развитием нефротического синдрома.
Синдром Барракера-Симонса
Болезнь наследуется по рецессивному типу и проявляется прогрессивным уменьшением подкожной жировой клетчатки в области лица, шеи и плечевого пояса. Нефротический синдром рефрактерен к терапии и сопровождается быстрым развитием ХПН. При морфологическом исследовании обнаруживают мембранопролиферативный гломерулонефрит. Болезнь развивается в детском возрасте. Возможны другие клинические варианты течения в виде гематурии или низкой протеинурии.
Врожденный сифилис
Одной из причин врожденного нефротического синдрома является врожденный сифилис. Протеинурия появляется сразу после рождения и спустя 2-3 мес развивается нефротический синдром. При морфологическом анализе выявляется мембранозный гломерулонефрит с выраженным тубулоинтерстициальным компонентом, иногда с явлениями мезангиальной пролиферации. У больных наблюдается также гепатоспленомегалия, диффузная инфильтрация кожи или пузырчатка, реже — поражение ЦНС и костной системы. Прогноз нефротического синдрома благоприятный при своевременной антибактериальной терапии (Папаян А.В., Стяжкина И.С., 2002).
Синдром приобретенного иммунодефицита при инфицировании ВИЧ в перинатальном периоде
Нефротический синдром развивается на 3-4-м месяце жизни. При морфологическом исследовании выявляется фокально-сегментарный гломерулосклероз, реже - мезангиопролиферативный гломерулонефрит. Нефротический синдром характеризуется быстрым прогрессированием с развитием ХПН.
Врожденный токсоплазмоз
Нефротический синдром может развиваться в первые 2-3 мес после родов или регистрироваться при рождении. Гистологически варианты поражения почек разнообразны, описаны фокально-сегментарный гломерулосклероз, мезангиопролиферативный гломерулонефрит с или без тубулоинтерстициального компонента. Нередко протекает с поражением глаз, центральной нервной системы.
Нефротический синдром при фибриллярном и иммунотактоидном гломерулонефрите
Фибриллярный и иммунотактоидный гломерулонефриты являются редкими разновидностями гломерулопатий. Описан случай ВИЧ-ассоциированного иммунотактоидного гломерулонефрита. Нами наблюдалось двое пациентов с фибриллярным гломерулонефритом. В обоих случаях развился нефротический синдром, быстро прогрессировала ХПН, и в итоге спустя несколько лет от дебюта заболевания был зафиксирован летальный исход. В целом однолетняя выживаемость, по некоторым данным, составляет 100%, 5-летняя — 80%. Морфологическими проявлениями служат гломерулярные депозиты в виде фибрилл и микротубул диаметром 12-32 нм, содержащие IgG и СЗ-комплемент. В отличие от амилоидных масс они не окрашиваются конго красным и триофлавином Т. При этом требуют исключения системные заболевания, гемобластозы, СД, при которых в редких случаях такие депозиты также могут иметь место.
Клинические проявления иммунотактоидного и фибриллярного гломерулонефрита не имеют специфических черт, отличающих их от других форм гломерулонефритов. Нефротический синдром является наиболее частым синдромом в клинической картине. Это было показано при анализе клиники 186 больных иммунотактоидным гломерулонефритом.
Обращает на себя внимание низкая эффективность иммунодепрессивной терапии. Удельный вес частичной ремиссии не превышает 9%, тогда как в 91% случаев позитивного эффекта от лечения не наблюдается. ХПН у 50% больных развивается уже на 2-4-м году болезни. В случае обнаружения при гистологическом исследовании конгонегативных субстанций, происходящих из иммуноглобулинов, дифференциальную диагностику проводят с криоглобулинемическим и волчаночным гломерулонефритом. Если эти депозиты происходят из внеклеточного матрикса, необходимо исключить врожденные и приобретенные гломерулопатии. Особую сложность представляет исключение врожденных гломерулопатии. Несмотря на появление гломерулопатии в детском возрасте, гистохимическое исследование на фибронектин проводится лишь в отдельных патоморфологических лабораториях. В случае конгопозитивных депозитов следует думать об амилоидозе, форма которого уточняется при проведении иммуногистохимического исследования биопсийного материала.
3. Патологические состояния, при которых нефротический синдром не встречается
Выделение данного раздела произошло по причине нередко возникающих заблуждений, приводящих к ошибочной диагностике. Нефротический синдром не встречается при остром постстрептококковом гломерулонефрите, поражении почек при системной склеродермии, пиелонефрите, интерстициальном нефрите, гипертоническом нефроангиосклерозе. Наиболее часто хронический гломерулонефрит, остро дебютирующий с нефротического синдрома, принимается за острый гломерулонефрит. При анализе нефробиоптата нередко морфологом дается заключение о наличии острого диффузного пролиферативного гломерулонефрита. Однако при наблюдении за этими больными у всех в дальнейшем диагностируется хронический гломерулонефрит. По нашим данным, при анализе нефробиопсий нефритов, дебютирующих острым нефротическим синдромом, морфологическая картина соответствует острому гломерулонефриту в 4% случаев (12 из 300 случаев) от всех гистологически идентифицированных гломерулонефритов. Однако наблюдение за этими больными подтвердило наличие у всех больных хронического гломерулонефрита.
ЛИТЕРАТУРА
1. Нефрология. Ключи к трудному диагнозу/ М.М.Батюшин – Элиста: ЗАОр НПП "Джанагар", 2007.
2. Нефрология. Основы диагностики / Под ред. дроф. В. П. Терентьева. (Серия "Медицина для Вас".) — Ростов н/Д: Феникс, 2003.
Похожие работы
... . Обычно поражение почек наблюдается в фазе анкилоза, когда постановка диагноза не представляет больших затруднений. Системная красная волчанка Нефротический синдром является частым проявлением люпус-нефрита. В 20% случаев заболевание дебютирует с нефротического синдрома, при этом в половине случаев кожные проявления, фотосенсибилизация, артрит, полисерозиты, характерные для системной красной ...
... В клинической практике наиболее часто встречаются такие причины развития нефротического синдрома, как диабетическая, паранеопластическая нефропатия и хронический гломерулонефрит. Для дифференциальной диагностики причин нефротического синдрома требуется в первую очередь информация, полученная при беседе с больным и анализе имеющейся медицинской документации, а также при объективном обследовании. ...
... , аминогликозиды), а также восстановление почечной функции после отмены данных лекарств позволяет произвести дифференциальную диагностику. Далеко не все патологические состояния сопровождаются развитием отеков, однако их появление на фоне хронической сердечной недостаточности объясняет их сочетание с отечным синдромом. Более того, в ряде случаев генез отечного и мочевого синдромов объясняется ...
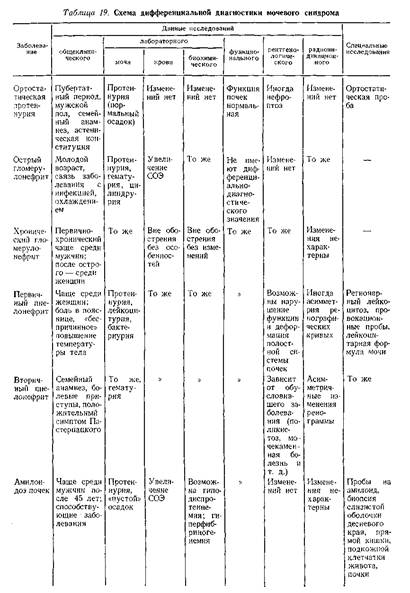
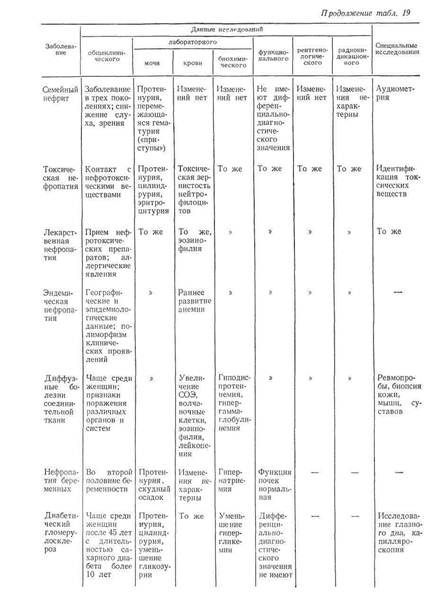
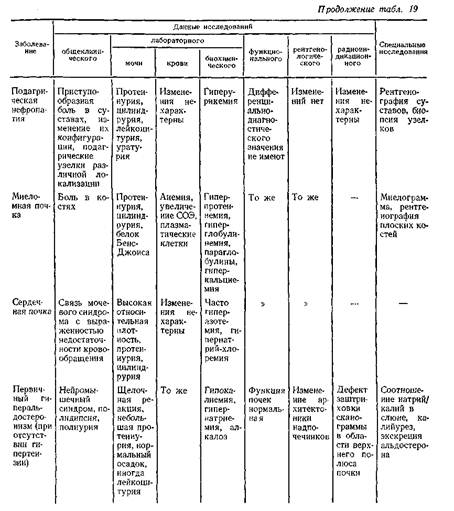
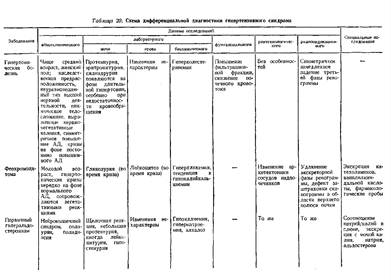
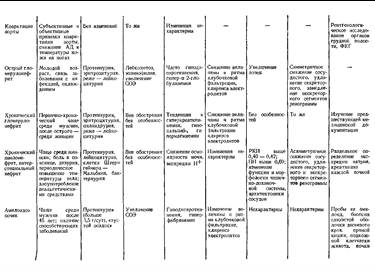
... заболеваниями соединительной ткани приходится с гломерулонефритом. О наличии сердечной почки свидетельствует положительная динамика мочевого синдрома в связи с уменьшением недостаточности кровообращения. Возможно сочетание гломерулонефрита и сердечной почки. ГИПЕРТЕНЗИВНЫЙ СИНДРОМ ПРИ ЗАБОЛЕВАНИЯХ ПОЧЕК Артериальная гипертензия является одним из самых частых симптомов при заболеваниях почек. ...
0 комментариев