Реферат на тему:
«Роль оксида азота как медиатора воспаления и фактора атерогенеза»
Роль оксида азота как медиатора воспаления и фактора атерогенеза
N0 является одним из важнейших медиаторов широкого спектра гомеостатических функций. Он продуцируется соответствующей синтетазой (N05), которая существует в 3 различных изоформах - нейрональной (п!ЧО8, тип 1), индуцируемой (ШОЗ, тип 2) и эндотелиальной (еМО8, тип 3). Изоформа пМО5 принимает участие в регуляции активности симпатической нервной системы, угнетает ее тонус и выраженность влияния на сердце и стенку сосудов [291]. Экспрессия пМО8 отмечена и за пределами центральной и периферической нервной систем, и N0, продуцируемый периваскулярными нервами на мозговых артериях, прямо модулирует их тонус. С помощью иммунных методов было показано, что пМО8 экспрес-сируется также в эндотелиоцитах, в сосудистых ГМК и в кардиомиоцитах, где она компартментализована в саркоплазмати-ческом ретикулуме и осуществляет аутокринную регуляцию функции клеток. Ее активация в кардиомиоцитах сопровождается увеличением скорости расслабления и угнетением сократимости, как базальной, так и стимулированной изопротеренолом. Напротив, кардиомиоциты мышей с генетическим отсутствием пМО8 характеризуются более высокой сократимостью, замедленным расслаблением и усилением сократительного ответа на стимуляцию (3-адренорецепторов [14].
В пределах сердечнососудистой системы ген е>Ю8 экспрессируется эндотелиоцитами, кардиомиоцитами и тромбоцитами. Наиболее мощным активатором экспрессии еМО5 в эндотелии является ЛФХ, который содержится в окисленных ЛПНП. При обработке изолированных эндотелиоцитов пупочной вены человека ЛФХ экспрессия мРНК еМО5 возрастает максимально в 11 раз, увеличивается содержание белка еМО5, его активность и продукция N0. Поэтому у кроликов на начальных этапах атеросклероза в эндотелиоцитах обнаруживается более высокое, чем в норме, содержание белка е!ЧО8. Статины умеренно повышают уровень
Ген 1КО5 экспрессируется практически всеми ядерными клетми сердечнососудистой системы, включая эндотелиальные, докардиальные, кардИОМИОЦИТы и ГМК, но прежде всего - воспалительными клетками субэндотелиального пространства (лейкоцитами, фибробластами и тучными клетками). Экспрессия 11ЧО5 в клетках макрофагального ряда и ГМК определяет их цитотоксическое действие, участие в иммунном ответе и запускается цитокинами (ФНО-а, ИЛ-1, ИЛ-2, 1Р1Ч-у) и ЛПС. Продолжительность жизни мРНК 1МО8 в изолированных макрофагах составляет примерно 3 ч.
Функциональная значимость N0, продуцируемого разными изоформами N08, различна. Так, через 1—3 сут после окклюзии срединной церебральной артерии размер зоны ИМ и нейрологические нарушения значительно меньше у мышей, лишенных 1тМО8. В то же время, введение им ингибитора ?N08 (Ь^АМЕ) увеличивает размер поражения. Это означает, что N0, продуцируемый пМО5, увеличивает тяжесть поражения при ишемии мозга, е!ЧО8 - уменьшает.
Существенно отличная функция присуща N0, который продуцируется еNО8. Он является одним из важнейших факторов регуляции структуры и функции стенки сосуда, обладает способностью осуществлять ЭЗР, угнетать сократительную и митогенную активность ГМК, подавлять адгезивность эндотелиоцитов и клеток крови — тромбоцитов и лейкоцитов. Ингибиторы еNО8 нарушают реактивность стенки сосуда, повышают АД, ослабляют кровоток, усиливают адгезию клеток крови к эндотелию, способствуют развитию локальных воспалительных явлений. Помимо этого, снижение активности еNО8 сопровождается усилением локальной продукции в стенке сосуда А II, который в еще большей степени уменьшает биодоступность N0 посредством активации NАРН-оксидазы эндотелиоцитов, моноцитов и ГМК с увеличением продукции СОР. Поэтому применение ингибиторов АПФ способно восстанавливать активность еNО8 в стенке сосуда. У животных, лишенных гена еNО8, отмечают повышение АД, Умеренную легочную гипертензию, резкое возрастание содержания ренина в крови. N0 является также регулятором ремоделирования сосудов в ответ на изменения потока крови, и у мышей с отсутствием гена еМО8 отмечают возрастание толщины стенки за счет выраженной пролиферации интимы. В эндотелиоцитах, регенерирующих после проведения ангиопластики, происходит усиленная экспрессия еМО5, и содержание N0 возрастает в 6 раз, существенно ограничивая образование неоинтимы и ремоделирование стенки сосуда. Угнетение еМО5 в этих условиях с помощью Ь-КАМЕ сопровождается трехкратным усилением экспрессии ЭТ-1 и ТСР-3 в эндотелиоцитах, возрастанием активности АПФ и образования А II, которые обладают как хемоаттрактантной, так и митогенной активностью в отношении ГМК с последующим развитием рестеноза [232].
Помимо этого, через N0 реализуется действие сосудистого эндотелиального фактора роста (УЕСР), и блокаторы еМО5 угнетают миграцию, пролиферацию эндотелиоцитов и ангиогенез. Поэтому у мышей с отсутствием еКО5 хроническая ишемия конечности не вызывает ангиогенной реакции и увеличения количества капилляров, как у нормальных животных, и экзогенное введение УЕОР не восстанавливает ангиогенез.
Изоформа 1МО5, индуцируемая цитокинами в эндотелиоцитах, ГМК и макрофагах, продуцирует в 100—1000 раз больше N0 по сравнению с постоянно экспрессированной еМО5. В малых концентрациях N0 является одним из важнейших физиологических регуляторов, тогда как в высоких он становится медиатором воспалительной реакции и оказывает выраженное цитоток-сическое действие. N0 быстро связывается с гемоглобином, который переводит его в неактивные продукты - нитриты и нитраты, а также взаимодействует с СОР с образованием ПОН и других мощных оксидантов, обладающих высокой цитотоксической активностью и принимающих участие в повреждении органов тканей и нарушений их функции. В зоне пораженного миокарда при ИМ, миокардите и сепсисе избыточная продукция N0, вызванная экспрессией 1ТМОЗ в инфильтрировавших макрофагах, является причиной дисфункции, повреждения и апоптоза кардиомиоцитов.
Изоформа 1МО5 при адекватной стимуляции может индуцироваться во многих клетках, приводя к интенсивному высвобождению N0, который способен как вызывать апоптоз, так и защищать клетки от апоптоза. Первый эффект связан с его свойствами При ишемии в миокарде отмечают достоверное уменьшение экспрессии мРНК еNО5 и возрастание - мРНК ^NО8; аналогично изменяется содержание в миокарде белка еNО8 и {N08. В условиях ишемии параллельно возрастанию экспрессии ^NО8 происходит повышение уровня нитритов, и оба эти эффекта быстро исчезают при реперфузии. Источником N0 при ишемии являются как мигрировавшие клетки крови, так и резидентные макрофаги и тучные клетки, которые способны дегранулировать и высвобождать ФНО-а, запускающий экспрессию {N08, повреждение миокарда и эндотелиоцитов. Применение симвастатина на изолированном сердце в условиях 15 мин ишемии и 3 ч реперфузии сопровождалось сохранением экспрессии мРНК ?N08, предупреждением возрастания экспрессии мРНК и белка ^NО5, уменьшением суммарной продукции нитритов, сохранением структуры и функции кардиомиоцитов и клеток эндотелия. Эти эффекты определялись предупреждением угнетения еNО8 и уменьшения продукции ею N0, так как параллельное применение Ь-МАМЕ полностью устраняло защитное действие симвастатина [191].
N0, продуцируемый {N08, оказывает также прямой отрицательный инотропный и цитотоксический эффекты на кардиомиоциты и играет существенную роль в генезе дисфункции и ремоделирования сердца. Установлена выраженная экспрессия {N08 в миокарде пациентов с ИБС, дилатационной кардиомиопатией (ДКМП), сопровождающаяся угнетением сократимости сердца, тогда как применение селективных ингибиторов {N08 позволяет существенно нормализовать его функцию. Особенно это значимо в связи с тем, что индукция {N08 в условиях воспаления может происходить непосредственно в кардиомиоцитах в результате активации НР-кВ провоспалительными цитокинами. Показано, что кардиомиоциты мышей способны индуцировать N08 и высвобождать N0 под действием цитокинов типа 1РМ-у и ИЛ-1(3 или ФНО-а и ИЛ-6 в присутствии ЛПС. Этот эффект наблюдали также у мышей с экспериментальным иммунным миокардитом [161]. О значимости 11ЧО8 в нарушениях структуры и функции миокарда в этих условиях свидетельствует то, что у мышей с отсутствием его гена развитие ИМ характеризуется менее выраженными нарушениями кардиодинамики, гибелью кардиомиоцитов в пери-инфарктной зоне, меньшей частотой летальных исходов. Помимо этого, нарушения сократительной функции кардиомиоцитов, возникающие при их инкубации с ИЛ-1(3, сочетаются с пропорциональным возрастанием продукции N0.
Активирующий эффект противовоспалительных цитокинов на индуцирование 1КО8 и синтез N0 существенно возрастает при одновременном действии СРП, что в определенной степени объясняет его роль в определении тяжести исхода ИМ [123].
Экспрессия 11ЧО8 значительно возрастает при различных иммунных, острых и хронических воспалительных реакциях. В ряде случаев это оказывает защитный эффект в результате антимикробной и противоопухолевой активности N0. В частности, при инфицировании нормальных мышей бактериями Ымепа в значительной части случаев отмечали спонтанное выздоровление, тогда как у гомозиготных мышей с отсутствием 11ЧО8 закономерно возникало выраженное поражение внутренних органов, и животные погибали.
Однако в других условиях N0 может оказывать повреждающее действие, так как он принимает участие в развитии локальных воспалительных реакций, сепсиса и формировании отека. Так, у мышей, лишенных 1МО8, локальный некроз кожи стопы вызывал значительно меньший отек, но введение подобным животным ЛПС сопровождалось более выраженной адгезией и прокатыванием лейкоцитов в посткапиллярных венулах. Это свидетельствует о том, что продуцируемый 1МО8 N0, наряду со значимостью одного из важнейших медиаторов воспалительной реакции, является ингибитором трансэндотелиальной миграции лейкоцитов и может оказывать противовоспалительное действие.
В ряде исследований показано, что направленность эффекта активации 11ЧО8 и усиления продукции N0 в значительной мере определяется ее источником. Так, при инкубации с активированными нейтрофилами в кардиомиоцитах мышей возрастала интенсивность оксидантного стресса в сочетании с быстрым угнетеиных нейтрофилов было связано с действием N0 и не наблю-р°восьПри инкубации с нейтрофилами животных с генетическим Д гутствием 1МО8. При этом активность 11ЧО8, экспрессируемой посредственно в кардиомиоцитах, оказывала защитное действие, способствовала сохранению реактивности по отношению к инотропным влияниям. В реальных условиях нейтрофилы, мигрировавшие через стенку сосуда, экспрессируют цитокин сс4-ин-тегрин и через него адгезируют к кардиомиоцитам, способствуя развитию в них оксидантного стресса и нарушению функции. Этот эффект возникает сопряженно с усилением экспрессии 1МО8 в нейтрофилах и, возможно, является его отражением [215].
Патогенетическая роль индукции 1МО8 в сосудистых ГМК особенно значима при сепсисе, когда она является причиной сосудистого коллапса, а угнетение 11ЧО8 в этих условиях значительно улучшает гемодинамику и снижает частоту гибели животных. У нормальных мышей в течение 5 ч после введения ЛПС наблюдали резкое падение АД и гибель, тогда как у гетерозиготных мышей, лишенных гена 11ЧО8, АД снижалось только на 30 %, у гомозиготных - на 15 % при отсутствии летальных исходов. Повышенная резистентность к действию ЛПС у мутантных мышей определялась уменьшением содержания в крови N0, так как уровень ФНО-а, ИЛ-1 и ИЛ-6 был аналогичным у животных обоих генотипов.
То, что гипотензия, вызываемая эндотоксином, связана с активацией 1МО8 и избыточной продукцией N0, подтверждается способностью аминогуанидина, селективного ингибитора ^N08, предупреждать как развитие гипотензии, так и возрастание уровня нитритов/нитратов в плазме. Предполагают, что усиленная индукция 1МО8 в условиях эндотоксинового шока является результатом действия ФНО-а, так как уменьшение его продукции с помощью активированного протеина С, физиологического антикоагулянта, сопровождается снижением активности 1М08, Уменьшением высвобождения N0 и предупреждением развития гипотензии [125].
Установлено также, что критическое падение давления в условиях геморрагического шока, возникающего при острой кровопотере и приводящего к летальному исходу, связано не только с гиповолемией, но и с активацией фактора NР-кВ и развитием каскада воспалительных явлений. При этом резко возрастает со модифицированную форму. Помимо этого, при реакции ПОН с СО2 образуется еще более мощный нитрирующий продукт, предположительно нитрозопероксокарбонат ((ЖООСО2").
Одним из механизмов повреждающего действия ПОН является способность высвобождать из антиоксидантного фермента церулоплазмина ионы меди с последующим проявлением присущего им широкого спектра прооксидантного действия — превращения СОР в гидроксильный радикал, каталитического участия в оксидантной модификации ЛПНП, особенно их гликозилиро-ванной формы, образования гидроксильного радикала при взаимодействии с остокоферолом, участия в образовании цероида макрофагами в присутствии аскорбиновой кислоты. Поэтому высокое содержание в сыворотке ионов меди (более 1,02 мл/л) или церулоплазмина, которое обычно отмечают у лиц с сосудистой патологией, особенно при нестабильной стенокардии, сочетается возрастанием риска ИМ более чем в 3,5 раза [77].
В последние годы установлен и альтернативный путь образования реактивных соединений азота с участием МРО — белка, синтезируемого в нейтрофилах и моноцитах, и находящегося в большом количестве в атеросклеротически пораженной сосудистой ткани. МРО может образовывать нитрирующие промежуточные продукты, которые превращают ЛПНП в модифицированную форму, захватывающуюся макрофагами. Помимо этого, ЛПНП, выделенные из поражения, обогащены хлортирозином - специфическим продуктом реакции между МРО и гипохлористой кислотой (НОС1). Антитела к нитрохлортирозину распознают эпитопы в атероме, локализация которых совпадает с локализацией мононуклеарных фагоцитирующих клеток. Показано, что МРО также может использовать нитрит (КО2") - главный конечный продукт метаболизма N0, как субстрат для нитрирования тирозиновых остатков белков и инициации ПОЛ. ЛПНП, подвергшиеся действию системы (МРО—Н2О2—МО2), быстро модифицируются и превращаются в форму, которая интенсивно захватывается макрофагами с образованием пенистых клеток [101].
В пересаженном сердце закономерно отмечают ускоренную динамику атеросклеротического поражения венечных артерий. Она в значительной мере определяется развитием иммунного воспаления и опосредована вовлечением в реакцию 1МО5 и ЦОГ-2, та как продуцируемые ими N0 и простаноиды обладают проатерО' генным действием. Между двумя этими ферментными системами существуют перекрестные взаимоотношения, и N0 в высокой концентрации повышает активность ЦОГ-2, в результате чего продуцируются провоспалительные простаноиды и СОР. Как при нативном, так и трансплантационном атеросклерозе 1М05 и ЦОГ-2 характеризуются совместной локализацией, преимущественно — в макрофагальных пенистых клетках. Экспрессия ЦОГ-2 установлена также в ГМК и эндотелиоцитах, включая эн-дотелиоциты уаад Vа5о^ит. Окрашивание нитротирозина как маркера образования и выраженности прооксидантного действия ПОН на тирозиновые группы белков также совпадает по локализации с экспрессией ЦОГ-2 и 11ЧО5. Действие ЦОГ-1 и еМО5 по многим параметрам рассматривается как антиатерогенное, а эффекты ЦОГ-2 и 1КО8 обладают проатерогенной направленностью в связи с высокой интенсивностью продукции и высвобождения простаноидов и N0.
Значимость индуцируемой ЦОГ-2 и продуктов ее активности в определении характера клинического течения ИБС была подтверждена в исследовании, включавшем 5529 пациентов с ИБС, которым проводили терапию аспирином. В течение 5 лет наблюдения в каждом последующем квартиле содержания в моче ТхВ2 установлено достоверное возрастание риска развития конечных кардиальных точек, соотношение величин которого при сопоставлении показателя в верхнем и нижнем квартилях достигало для ИМ составляло 2, для коронарной смерти - 3,5.
Активность тромбоцитарной ЦОГ-1, продуцирующей РОН2 ~ предшественник ТхА2, полностью и необратимо угнетается аспирином в дозе 80—325 мг в сутки. Однако индуцируемая ЦОГ-2, экспрессируемая клетками стенки сосуда, значительно менее чувствительна к аспирину, и для ее блокады необходимо не менее 500 мг аспирина в сутки. Поэтому даже в-условиях блокады ЦОГ-1 8 тромбоцитах сосудистая ЦОГ-2, экспрессия которой под влиянием провоспалительных стимулов возрастает в 10-20 раз, продолжает синтезировать РОН2, из которой в тромбоцитах УЩествляется синтез ТхАз- Это во многом определяет резистен-ность к аспирину и высокий риск развития острых коронарных х° Ытий у пациентов, у которых на фоне приема аспирина со-Раняется высокий уровень синтеза ТхА2 [67]. ЦогЗНаЧальнс> предполагали, что постоянно экспрессируемая продуцирует «физиологические» простагландины, тогда
как индуцируемая ЦОГ-2 — провоспалительные. Однако в настоящее время уже четко установлено, что ЦОГ-2 также имеет выраженные физиологические функции и постоянно экспрессирова-на в эндотелии; ее угнетение при применении аспирина в высоких дозах устраняет свойственные эндотелиоцитам анти-тромбоцитарный и антитромботический эффекты и приводит к развитию протромботического состояния в связи с резким угнетением синтеза простациклина. В частности, у крыс с гипертензией селективное угнетение ЦОГ-2 приводило к росту АД и усиленной адгезии лейкоцитов к сосудистому эндотелию [187]. В малых дозах аспирин угнетает преимущественно ЦОГ-1, тогда как ЦОГ-2 продолжает продуцировать простациклин, подавляя активность тромбоцитов.
В отличие от доминировавших ранее представлений, условия воспаления и ГХЕ характеризуются скорее возрастанием, чем уменьшением продукции N0 [175]. Ослабление дилататорной функции эндотелия при действии цитокинов, окисленных ЛПНП, при атеросклеротическом поражении в большей степени связано со снижением биодоступности N0, чем с уменьшением суммарной его продукции и высвобождения [188]. Оно отражает инактивацию N0 усиленно продуцирующимся СОР, источником которого является преимущественно НАОРН-оксидаза мембран ГМК, эндотелиоцитов и воспалительных клеток. Так, через 18 ч после внутрибрюшинного введения крысам ЛПС в дозе 10 мкг/кг продукция СОР в изолированных сегментах аорты возрастала в 3 раза, перекиси водорода — в 2 раза. Эти эффекты сочетались с увеличением в 2-3 раза экспрессии тКЛМА и белка NА^РН-ок-сидазы и ксантин оксидазы. Параллельно происходило усиление ПЧО8-зависимой продукции N0, а взаимодействие СОР и N0 в эквимолярных концентрациях сопровождалось образованием ПОН, который и определял дисфункцию эндотелия с выраженной активацией ЫР-кВ и возрастанием экспрессии Е-селектина.
При активации лейкоцитов происходит резкое увеличение продукции СОР, и в этом случае доступность N0 является фактором* ограничивающим образование ПОН. Показано, что повреждение эндотелиоцитов при инкубации с активированными нейтрофила ми обусловлена главным образом действием ПОН, и блокада еN значительно его уменьшала и ограничивала нитрирование пР°те нов, тогда как донаторы N0 усиливали эти эффекты. Помимо э го, лейкоциты способны прямо высвобождать ПОН, так как в нерофилах и моноцитах при активации ЛПС индуцируется {N08 и продуцируется значительное количество N0 [251].
В условиях локального воспаления также отмечают усиленную экспрессию в стенке сосуда 1МО8 и резкое возрастание суммарной продукции N0, сочетающееся с угнетением еМОЗ и уменьшением образования ею N0. Эти изменения возникают при действии провоспалительных цитокинов (ИЛ-1, ФНО-а, 1РМ-у), бактериального ЛПС, модифицированных ЛПНП. Экспрессию тКЛМА и белка 1МО8 отмечают почти исключительно в клетках интимы и медии, и удаление эндотелия практически не отражается на ее выраженности. Установлено, что инкубация культуры ГМК в среде с добавлением с ИЛ-1(3 или ФНО-а сопровождалась высвобождением N0 и сопутствующей цитотоксичностью, и этот эффект предупреждался ингибиторами 11ЧО8. Помимо этого, ФНО-а способен индуцировать экспрессию 1МО8 в макрофагах, что обусловливает его ведущую роль как киллера патогенных агентов. Продукция больших количеств N0 в этих условиях, благодаря его цитотоксическому и антибактериальному действию, является важным звеном иммунорезистентности.
Возрастание активности 11ЧО8 в макрофагах, ГМК и эндотелиоцитах под влиянием провоспалительных цитокинов сочетается с более чем трехкратной активацией синтеза тетрагидробиоптерина — кофактора ПЫО8, содержание которого в культуре клеток эндотелия пупочной вены человека при сочетанной стимуляции 1РК-у, ФНО-а и ИЛ-1 возрастает более чем в 140 раз. В отсутствие тетрагидробиоптерина N08 продуцирует не N0, а СОР, и применение тетрагидробиоптерина при оксидантном стрессе, ГХЕ, курении восстанавливает продукцию N0. При этом уменьшение биодоступности тетрагидробиоптерина может быть следствием °ксидантного стресса и происходить под действием ПОН.
Неоднократно подтверждено, что между активностью еNО8 и !^О8 существует реципрокная зависимость. Поэтому удаление эндотелия или нарушение его функции способствует появлению СТСНКесосУда активности ^NО8, так как N0, продуцируемый ^08 в эндотелиоцитах, подавляет экспрессию {N08 в сосудис-1х ГМК. В исследовании, проведенном на сонной артерии кры-*' показано, что через 7—30 дней после удаления эндотелия в ц ^ отчетливо возрастала экспрессия ^NО8, а у крыс с тромбо-ьт,,„ОПениеиона возникала раньше и была значительно более кенной. Уменьшение экспрессии [N08 и продукции N0 под действием тромбоцитов в условиях деэндотелизации связано с тем, что РООР является естественным ингибитором {N08, и тромбоцитопения или блокада рецепторов тромбоцитов (трапидил, Рео-про) способствуют усилению продукции N0 1КО5 и предупреждают развитие неоинтимы [132]. Напротив, усиленная экспрессия 1МО8 в клетках стенки сосуда при атеросклерозе и ГХЕ и возрастание суммарной продукции N0 сопровождаются по принципу отрицательной обратной связи угнетением активности еМО8 и нарушением ЭЗР. Показано, что как сам N0, так и его экзогенные донаторы, угнетают е!ЧО8, тогда как инактиватор N0 оксигемоглобин повышает ее активность. Кроме того, одним из эффектов N0 в высокой концентрации является угнетение цитохрома Р-450 в эндотелиоцитах и последующее уменьшение синтеза эндотелийзависимого фактора гиперполяризации, который определяет вазомоторную активность эндотелия, прежде всего резистивных сосудов.
Однако регуляторное влияние N0 на индукцию гена 11ЧО8 отличается в клетках различного типа. В макрофагах он уменьшает экспрессию белка 1МО8 вследствие угнетения фактора КР-кВ, и наличие в макрофагах отрицательной обратной связи по N0 обеспечивает механизм, контролирующий количество продуцируемого N0 и предупреждающий его токсическое действие. В то же время, в ГМК N0 не влияет на индукцию гена 11ЧО8, что объясняет стабильно высокую продукцию N0 при септическом шоке, приводящую к развитию острой сосудистой недостаточности.
В то же время, интегральное действие 11ЧО8, по мнению ряда авторов, можно рассматривать как защитное антиатерогенное, так как оно способствует поддержанию гомеостаза ХС. В частности, у мышей с генетическим дефицитом 11ЧО8 системное АД, как и содержание ХС в крови и частота выявления атером в стенке аорты, значительное выше, чем у нормальных животных того же возраста (3, 6, 12 мес.). Предполагают, что этот эффект реализуется через способность N0 предупреждать пероксидацию ЛПНП, угнетать адгезию тромбоцитов, агрегацию лейкоцитов и их взаимодействие с эндотелием [122].
Результаты исследований последних лет свидетельствуют о том» что в ряде патологических ситуаций активация 11ЧО8 с усиленной продукцией N0 оказывает защитный эффект и сочетается с угнетением отдельных механизмов воспаления и атеросклероза. ПреЖ" всего, антиатерогенное действие N0 связано с его способность
угнетать клеточную пролиферацию. Так, хроническая ингаляция N0 после баллонного повреждения сонной артерии у крыс уменьшает ТИМ через 2 нед на 43 %, а однократное применение донаторов N0 уменьшает выраженность гиперплазии интимы на 77 % через 2 нед после повреждения сосуда. На ранних этапах после трансплантации сердца активация 11ЧО8 предупреждает ремоделирование сосудов трансплантата, и ее угнетение сопровождается быстрым возрастанием толщины интимы и уменьшением сосудистого просвета [16]. Об этом свидетельствует в пять раз более выраженное возрастание ТИМ венечных артерий, вдвое большее содержание в интиме ГМК, вдвое большее уменьшение сосудистого просвета через 55 дней после пересадки сердца у мышей с генетическим отсутствием 1МО8.
Защитную роль 11ЧО8 и продуцируемого ею N0 для развития посттрансплантационного артериосклероза связывают с угнетением рекрутизации в стенку сосуда преимущественно провоспалительных ТЫ клеток, ослаблением миграции, пролиферации ГМК и образования ими коллагена. Помимо этого, N0 в высокой концентрации вызывает апоптоз макрофагов и ГМК, предупреждая пролиферативные процессы в неоинтиме. Поэтому ре-моделирование венечных сосудов пересаженного сердца у генетической линии мышей с дефицитом 1МО8 выражено в большей степени, чем у нормальных животных.
Связь между дисфункцией эндотелия и атеросклерозом во многом обусловлена устранением угнетающего действия N0 на пролиферацию ГМК. N0, а также все его донаторы способны угнетать пролиферацию ГМК, а в более высоких концентрациях вызывать их апоптоз. N0 также подавляет миграцию и пролиферацию ГМК, стимулированные А II, угнетает продукцию ГМК коллагена и фибронектина и индуцирует апоптоз макрофагов. Показано, что трансфекция гена 1МО8 в стенку артерии после ангиопластики с помо-Щью ретровируса приводила к значительному возрастанию синтеза N0, ослаблению пролиферации ГМК и уменьшению выраженности стенозирования, а применение ингибиторов 1МО8 сопро°Жцалось усиленной пролиферацией клеточных элементов инимы и возрастанием ее толщины более чем на 57 %.
Способность 11ЧО8 предупреждать гиперплазию клеточных элеНтов интимы и ремоделирование стенки в месте повреждения 1Хг/^а подтверждена также при трансфекции человеческого генаь помощью аденовируса в баллонированную артерию свири этом эффективность имплантации достигала только 1 %, но продукция нитритов возрастала в 100 раз, полностью угнетая образование неоинтимы. Таким образом, даже очень незначительная дополнительная экспрессия гена 1МО8 существенно модулирует сосудистый ответ на повреждение.
Процесс ремоделирования стенки сосуда при повреждении включает ряд последовательно развивающихся реакций. Прежде всего, происходит агрегация и адгезия тромбоцитов к эндотелию в сочетании с секрецией цитокинов, факторов роста и хемотаксисом лейкоцитов. Тромбоцитарный фактор роста (РВСР) запускает миграцию ГМК из медии в интиму, фибробластический фактор роста (РРСР) определяет первый этап их пролиферации после повреждения, а трансформирующий фактор роста (ТОР-(3) стимулирует превращение ГМК в секреторный фенотип с продукцией коллагена, что приводит к образованию внеклеточного матрикса. Пролиферация ГМК отмечается уже через 24 ч после повреждения и продолжается не менее 2 нед. Миграция ГМК в интиму обеспечивается 1-РА, который активирует ферменты, в частности — ММРз, разрушающие матрикс. Благодаря этому ГМК на 1—3-е сутки после повреждения проникают в интиму, где происходит их пролиферация. Усиленный синтез УЕСР обеспечивает регенерацию эндотелия, которая отмечается через 24 ч и длится на протяжении 6-10 нед. Сочетание пролиферативного процесса и усиленной продукции матриксных белков завершается утолщением интимы, которому противостоит ремоделирование сосуда, приводящее к увеличению его диаметра.
Все эти процессы в нормальных условиях находятся под постоянным регуляторным влиянием N0, который продуцируется сосудистым эндотелием и препятствует адгезии тромбоцитов. Однако при повреждении эндотелия основным источником N0 в стенке сосуда становится ПЧО8, который экспрессируется в ГМК, и применение в этих условиях ее ингибитора увеличивает в 3 раза адгезию тромбоцитов, в 15 раз - лейкоцитов и на 25 % уменьшает скорость кровотока. Параллельно индукция 1МО8 с высвобождением N0 препятствует адгезии моноцитов, активации НР-кВ и экспрессии гена УСАМ-1. Помимо этого, в тромбоцитах при активации также экспрессируется ПЧО8 и высвобождается N0, что подавляет их избыточную агрегацию.
Помимо этого, прогрессирующая на протяжении 2 нед сосудистой деэндотелизации экспрессия мРНК 1МО8 как в ГМК из медии в интиму и выраженность пролиферативных процессов, а также стимулирует миграцию эндотелиоцитов, их рост и ангиогенез, и подавляет апоптоз. На этом основании было сделано заключение, что экспрессия 1МО8 компенсирует потерю активности е!ЧО8 в результате разрушения эндотелия и потому является защитным механизмом, так как ослабляет рекрутирование моноцитов и нейтрофилов и ограничивает воспалительный процесс, Усиление экспрессии 1НО8 в миокарде определяет также защитный эффект так называемого «прекондиционирования» -уменьшения выраженности поражения и некроза миокарда в условиях стойкого прекращения его кровоснабжения. Состояние «прекондиционирования» возникает после предшествующей кратковременной ишемии как самого сердца, так и различных внутренних органов [264]. Показано, что трехминутная окклюзия верхней мезентериальной артерии, предшествующая на 24 ч 30-минутной полной окклюзия венечной артерии, приводила к уменьшению размера зоны риска от 72 до 44 %, зоны некроза - от 31 до 23 % параллельно со снижением активности МРО в миокарде как маркера инфильтрации лейкоцитов. Введение аминогуанидина — ингибитора ПЧО8 - устраняло как защитный эффект прекондиционирования, так и связанного с ним угнетения инфильтрации нейтрофилов [287].
Определено, что прекондиционирование как защитная реакция состоит из 2 фаз: ранней, которая развивается сразу после кратковременной ишемии, и поздней, возникающей через 24 ч и длящейся до 3 сут. Основным фактором поздней фазы прекондиционирования является N0, синтезируемый как е!ЧО8, так и 1МО8. Активация еМО8 является также ведущим механизмом ранней фазы прекондиционирования, длящейся до 3 ч и являющейся следствием действия брадикинина, который способен повышать активность е!ЧО8 более чем в 40 раз.
В то же время, у мышей с генетическим отсутствием е!ЧО8 Раннее прекондиционирование сохраняется, но значительно увеличивается длительность ишемии, необходимой для развития за-'Цитного эффекта. Это означает, что механизм раннего прекон-Диционирования включает, по-видимому, ряд факторов, в сочетании которых роль N0 заключается в снижении порога прекониционирования. В частности, к числу этих факторов относятся ^"8, которые образуются при различных стрессорных возтвиях, повышают устойчивость миокарда к действию ише и других тканях. Проведение на этом фоне АКШ сочеталось со значительным угнетением возникающей воспалительной реакции, повышением активности е>Ю8 и снижением - 11ЧО8, менее выраженным возрастанием концентрации ИЛ-6 и ИЛ-8 в плазме через 3 ч после вмешательства наряду с уменьшением тяжести поражения миокарда [100]. Индуцирование экспрессии Н8Р72 в изолированном сердце крысы гипертермией также оказывало существенное кар-диопротекторное действие, и воспроизведение через 24 ч ишемии и реперфузии на этом фоне приводило к достоверно более полноценному восстановлению функции сердца и менее выраженному высвобождению креатинфосфокиназы (КФК), чем в контроле [202].
Таким образом, N0, продуцируемый 11ЧО8, обладает многогранным действием — он участвует в реализации защитных реакций, которые запускаются цитокинами и активацией иммунной системы, обладает антибактериальной и противоопухолевой активностью, но может действовать и как эндогенный ингибитор защитных систем организма, оказывать цитотоксический эффект на нормальные ткани и вызывать клеточный апоптоз. Вопрос том, в какой степени функционирование 1МО8 в условиях воспаления и атеросклероза имеет защитный характер, а в какой способствует прогрессированию повреждения, остается спорным настоящее время, так как N0 в зависимости от концентрации и стимулов, вызывающих его продукцию, может иметь совершенно различные направленность и выраженность действия.
Похожие работы
... влияния статинов на апоптоз гладкомышечных клеток. Аторвастатин, симвастатин и ловастатин (но не правастатин, обладающий гидрофильными свойствами) могут вызывать апоптоз гладкомышечных клеток. Это действие статинов обусловлено их ингибирующим влиянием на изопреновую модификацию белков. Результаты исследования in vivoпоказали, что ловастатин, симвастатин и флувастатин (но не правастатин) ...
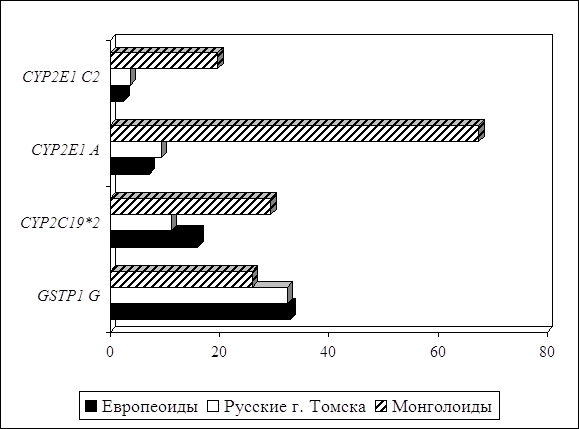
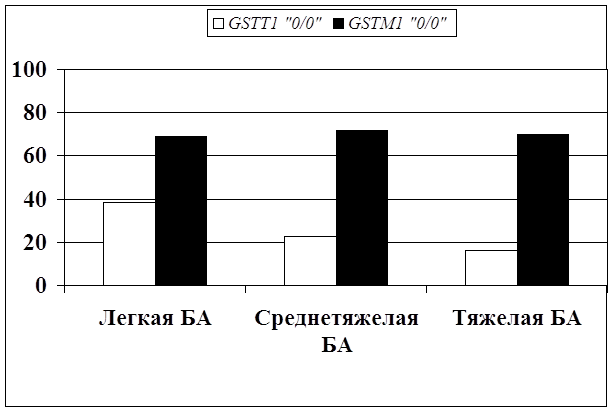
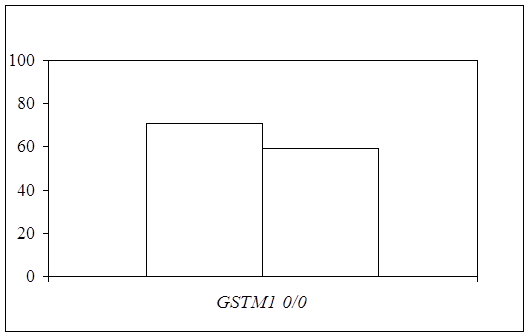
... препаратов. Установлена связь полиморфизма 313A>G гена GSTP1 с изменчивостью уровня аланинаминотрансферазы (р=0,021). 7. Выявлены различия в структуре генетической подверженности к бронхиальной астме и туберкулезу по генам ферментативной системы метаболизма ксенобиотиков: гены GSTM1, CYP2E1 и CYP2C19 связаны с бронхиальной астмой и значимыми для заболевания качественными и ...
... также следует учитывать при применении некоторых ЛС (например, снотворных длительного действия). Обилие лекарственных веществ, известных современной медицине, отнюдь не означает, что каждый препарат обладает индивидуальным механизмом действия. Понимание механизма действия важно не только для фармаколога, занятого поиском совершенных препаратов, но и помогает правильно использовать их в клинике. ...
... генетический анализ генов, кодирующих различные цитокины. В ходе такого анализа идентифицируются полиморфные варианты генов, кодирующих эти вещества. Активная работа в области изучения молекулярно-генетических механизмов псориаза и атеросклероза ведется и в лаборатории функциональной геномики Института общей генетики им. Н.И.Вавилова РАН. Здесь получили возможность оценить взаимосвязь этих двух з
0 комментариев