Резюме
Целью данного обзора является оценка имеющихся концепций индукции и ингибирования ферментов подсемейства CYP3A ксенобиотиками и другими химическими соединениями, с акцентированием роли ядерных рецепторов, мутаций экспрессируемых генов и их аллелей, внутривидовых вариаций, а также особенностей онтогенеза в этом процессе, поскольку в настоящее время все больше набирает вес утверждение, что имеется обширная база для взаимодействия среди членов семейства ядерных рецепторов, и причастности к вышеуказанному процессу стероидных рецепторов (подобных глюкокортикоидному рецептору (GR), и других факторов.
Введение
Фармакокинетическое взаимодействие лекарственных средств на этапе метаболизма возникает при использовании препаратов, способных изменять активность ферментов. Ксенобиотики, какой бы структурой они ни обладали, встречают на своем пути в организме адекватный фермент, переводящий их или в состояние, удобное для использования в качестве энергетического и пластического материала, или в состояние, удобное для выведения из организма. Система биотрансформации ксенобиотиков состоит из ряда ферментных систем, локализованных в межклеточном пространстве, на клеточных и субклеточных мембранах, внутри органелл клетки. При этом процессы биотрансформации весьма сложны и обычно включают ряд последовательных стадий, каждая из которых опосредуется определенным ферментом.
Ферментные системы окислительного метаболизма обнаруживаются практически у всех живых организмов, где они играют важную роль в детоксикации любых эндо - или экзогенных веществ. Из них самой важной и мощной является система цитохрома Р-450 (CYP) (термин "цитохром Р-450 " обозначает семейство изоферментов, которые сходны по физико-химическим, каталитическим и молекулярным свойствам.), которая рассматривается как наиболее универсальный биологический катализатор (так называемая "паяльная лампа"). Она объединяет мультигенное семейство гемопротеинов, ответственных за метаболизм многочисленных ксенобиотиков, включая терапевтические препараты, химикалии окружающей среды и диетические компоненты, а также эндогенные вещества, такие как жирорастворимые витамины, билирубин, тироксин, стероиды и желчные кислоты [Francois BERTHOU, Cytochrome P450 enzyme regulation by induction and inhibition,]. Никакая другая ферментная система не может соперничать с цитохромом P450 по диапазону метаболизируемых субстратов, различных по структуре и молекулярному весу – от метанола (MW=42), до циклоспорина А (MW=1203). Эта система представляет собой комплекс ферментов (цитохромы Р450 и b5, NADPH- и NADH-редуктазы и фосфолипидный компонент мембран) сосредоточенных в мембранах эндоплазматического ретикулума или других компартментах клеток. Основной компонент системы - цитохром Р450, представлен суперсемейством монооксигеназ смешанной функции (MFO), ответственных за I стадию окислительного метаболизма, которая во многом определяет пути биотрансформации ксеноботиков в организме [Н.И. Головенко, 1980; А.И. Арчаков, И.И. Карузина, 1988; Н.В. Адрианов, В.Ю. Уваров,1988; Lewis, Lake, 1997].
Цитохром Р450 (КФ 1.14.14.1) является типичным мембраносвязанным ферментом и большей частью своей молекулы погружен в липидный бислой, а каталитически активная гемовая часть расположена на границе раздела фаз. Проявление активности фермента возможно только в присутствии фосфолипидов или их аналогов, которые стабилизируют цитохром в функционально активной конформации [Н.В. Адрианов, В.Ю. Уваров,1988; В.Ю. Уваров и соавт., 1995; Dai et al., 1998; Yun et al., 1998]. Все цитохромы имеют консервативное структурное ядро и вариабельные участки в областях, вовлеченных в узнавание субстрата и связывание редокс-партнера [Peterson, Graham, 1998]. Особенности строения гидрофобного субстрат-связывающего кармана определяют субстратную избирательность фермента, поэтому, несмотря на значительную перекрестную специфичность, селективные субстраты и селективные ингибиторы отдельных цитохромов семейства P450 позволяют дать характеристику соответствующих его изоформ [Dai et al., 1998].["Effects of administration of cytochrome P450 inhibitors on the P450-mediated metabolism of organophosphorus compounds", Akua Agyeman, B.S., 1990, Seton Hall university]. Следует отметить, что из всех компонентов монокосигеназной системы только цитохром Р450 способен активировать молекулярный кислород, передавая ему электроны, полученные от NADPH и (или) цитохрома b5 [А.И. Арчаков, И.И. Карузина, 1988; Vaz et al., 1996; Coon et al., 1998]. Первый электрон поступает на цитохром Р450 от NADPH-редуктазы, второй - от цитохрома b5. В случае же окисления NADH оба электрона поступают от цитохрома b5. P450-катализируемая реакция моноокисления есть процесс, при котором один атом молекулярного кислорода реагирует с липофильным субстратом RH и в результате возникает гидрофобный метаболит ROH, вступающий в реакции коньюгирования с ферментами II стадии метаболизма. [Francois BERTHOU, Cytochrome P450 enzyme regulation by induction and inhibition,] (Рис.1).
Рис. 1 - Фазы окислительного метаболизма ксенобиотиков
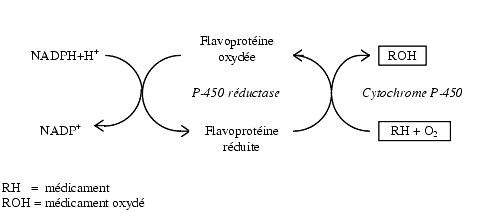
Сам механизм монооксигеназной реакции в общих чертах выглядит следующим образом. На первой стадии происходит связывание окисленной формы цитохрома Р450 с субстратом и образование фермент-субстратного комплекса (Fe3+RH), существование которого подтверждается тремя типами спектральных изменений. Большинство субстратов вызывают изменения 1-го типа за счет увеличения доли высокоспиновой формы фермента. На второй стадии идет восстановление электроном поступающим от NADPH-редуктазы с образованием восстановленного фермент-субстратного комплекса (Fe2+RH). Затем присоединяется молекулярный кислород и образуется оксикомплекс (Fe2+О2RH). На этой стадии может происходить разобщение комплекса в случае переноса электронов на кислород и после диссоциации комплекса возможно образование супероксидного анион-радикала. Для продолжения моноксигеназного цикла необходимо восстановление оксикомплекса вторым электроном и образование пероксикомплекса (Fe2+О2-RH). Затем идет гетеролитический разрыв связи О-О с освобождением воды и образованием комплекса (FeO)3+RH, в котором существует электронодефицитный (шестиэлектронный) оксеноидный атом кислорода. Дальнейший механизм окисления субстрата с участием оксена называется оксеноидным.
Возможны три пути взаимодействия оксеноидного комплекса с молекулой субстрата: прямое внедрение кислорода в связь С-Н; отрыв атома водорода от субстрата с образованием радикала субстрата и координированного с железом гидроксильного радикала с последующей рекомбинацией; отрыв гидрид-иона с промежуточным образованием карбониевого иона из молекулы субстрата. По какому конкретному пути пойдет реакция определяется строением субстрата и другими факторами. Оксеноидный комплекс не единственный окислитель, функционирующий в монооксигеназном каталитическом цикле. Такая роль приписывается и другим гипервалентным железо-кислородным комплексам - нуклеофильному пероксижелезу, нуклеофильному или электрофильному гидропероксижелезу, каждый из которых имеет свои пути взаимодействия с субстратом [Vaz et al., 1996, 1998; Coon et al., 1998]. Существование различных окислителей в каталитическом цикле обеспечивает вариабельность молекулярных механизмов окисления субстратов, широкую субстратную специфичность монооксигеназ, большое разнообразие продуктов реакции.
Монооксигеназные окислительные реакции повышают полярность гидрофобного субстрата, делают его доступным для действия ферментов конъюгирующих субстрат с глюкуроновой, серной, уксусной кислотами, другими веществами по появившимся или освободившимся в результате окисления полярным группам (II фаза метаболизма ксенобиотиков) [Н.И. Головенко, 1980; А.И. Арчаков, И.И. Карузина, 1988; K.W. Bock et al., 1990]. Конечные продукты окисления и конъюгации, как правило, обладают более низкой биологической активностью и в силу большей растворимости в воде быстрее элиминируются из организма. По отношению к большинству лекарственных веществ и ксенобиотиков техногенного происхождения это означает утрату ими фармакологической активности или токсичности, однако в ряде случаев образуются биологически активные метаболиты, как например, алкилирующие метаболиты многих цитостатиков, активные метаболиты анальгетиков, антиконвульсантов, антиаритмиков, токсические метаболиты канцерогенов, гепатотоксинов [Wolf et al., 1996]. Т.о., хотя монооксигеназные реакции по своей сути направлены на защиту живых систем от накопления гидрофобных соединений, они тем не менее представляет определенную опасность для организма, так как неотъемлемой частью каталитического цикла является образование промежуточных активных метаболитов, продуктов неполного восстановления кислорода, которые оказывают повреждающее действие, химически модифицируя макромолекулы, нарушая проницаемость мембран, стимулируя реакции перекисного окисления [А.И. Арчаков, И.И. Карузина, 1988; Chen, Cederbaum,1998; А.И. Арчаков и соавт., 1998].
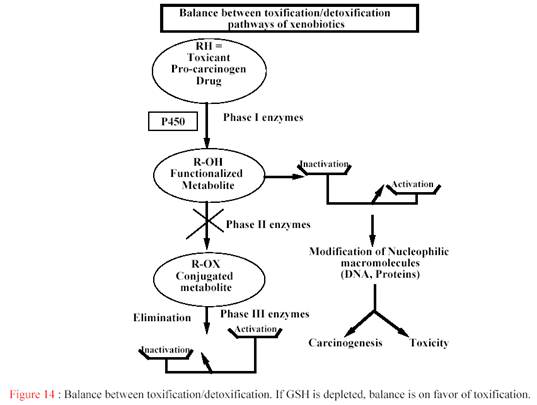
В ряде случаев особо агресивные свойства образующихся интермедиатов и недостаточная эффективность их обезвреживания служат причиной гепато- и нефротоксичности, мутагенеза, канцерогенеза, тератогенеза, аллергии и других проявлений химически индуцированной токсичности (Рис.20 ["Effects of administration of cytochrome P450 inhibitors on the P450-mediated metabolism of organophosphorus compounds", Akua Agyeman, B.S., 1990, Seton Hall university].
Рис. 2 - Баланс между токсификацией и детоксификацией
CYP суперсемейство существует в течение более чем 1,5 миллиарда лет (Рис.3) [3,4].
Рис. 3 - Онтогенез цитохрома P450 (CYP) 3A4 и 3A7 активности
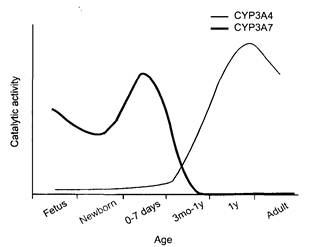
CYP система монооксигеназ, развившаяся, вероятно, в процессе совершенствования метаболизма стероидов как необходимая для сохранения мембранной целостности, впоследствии проявила себя как мощнейший фактор в дезактивации экзогенных соединений [5]. Считается, что все гены цитохрома Р450, от бактерий до человека, имеют общего предшественника, который появился более 2 млрд лет тому назад одновременно с появлением кислорода. В процессе эволюции происходило неоднократное разделение филогенетического дерева цитохрома Р450, что отражало состояние антагонизма между животными и растениями [Guengerich, 1992; Archakov, Degtyarenko, 1993; Lewis et al.,1998]. Наивысшие концентрации цитохрома Р-450 обнаруживаются в эндоплазматическом ретикулуме или микросомах гепатоцитов, меньшие – в энтероцитах тонкой кишки и еще меньшие – во внепеченочных тканях (почках, легких и мозге) [Gonzalez и другие., 1993; Pollock B. G. et al., 1999]. На сегодня обнаружено более 110 семейств цитохрома Р450, множество подсемейств и индивидуальных белков. Клонированы сотни генов различных цитохромов P450 у животных, растений, грибов и бактерий [Porter, 1995; Gonzalez, Lee, 1996]. Только у крыс их выделено более 50, а человека известно около 100 цитохромов P450, разделяемых на 17 семейств, кодируемых 48 генами и 10 псевдогенами. [Guengerich F.P., 1992; Beaune P., 1993; Horsmans, 1997; Ross A McKinnon, 2000], которые имеют значительные различия не только в структуре, вопросах активации и регуляции, субстратной специфичности итд, но и в аспектах онтогенеза.
[Saskia N.et all/Cytochrome P450 3A. Ontogeny and Drug Disposition, ClinPharmacokinetlW Dec: 37 (6);] Интересно, что растения имеют большее количество CYP генов, чем животные, из-за большего количества P450-зависимых критических путей метаболизма. Так, рекорд содержания цитохромов принадлежит маленькому растению Arabidopsis thaliana и равен 287, то есть приблизительно 1 % полного генома. [Francois BERTHOU, Cytochrome P450 enzyme regulation by induction and inhibition,]
В структуре всех изоформ цитохрома Р450 имеется консервативный участок вблизи СООН-конца, который содержит 26 аминокислотных остатков. В семейства включают белки, которые имеют около 36% подобия аминокислотного состава, в подсемейства - белки, аминокислотный состав, которых сходен на 65%. Внутри подсемейства белки имеют сходство более чем на 65%). [Akua Agyeman "Effects of administration of cytochrome P450 inhibitors on the P450-mediated metabolism of organophosphorus compounds", , B.S., 1990, Seton Hall university].
И до открытия цитохрома P450, было понятно, что эффективность многих лекарственных препаратов определяется скоростью их биотрансформации организмом. Активность лекарственных средств, метаболизирующихся ферментами печени заметно изменялась, когда животным дополнительно вводились гормоны, инсектициды и канцерогенные вещества – возникающая при этом ферментативная индукция ускоряла биотрансформацию лекарственных препаратов, уменьшая продолжительность и интенсивность их действия [Somogyi A, Kovacs K, Solymoss B, Kuntzman R, Conney AH. 1971. Suppression of 7,12-dimethylbenz(a)anthracene-produced adrenal necrosis by steroids capable of inducing aryl hydrocarbon hydroxylase. Life Sci. 10:1261–71]. Характер индукции ферментативной активности определялся типом индуктора, что свидетельствовало о существовании ряда ферментных систем, т.е. что большой спектр каталитической активности обеспечивается множественными ферментами, а не отдельный изоформой [Lu AYH, Somogyi A, West S, Kuntzman R, Conney AH. 1972. Pregnenolone-16-carbonitrile: a new type of inducer of drug-metabolizing enzymes. Arch. Biochem. Biophys. 152:457–62] [Bryan Goodwin, Matthew R. Redinbo, and Steven A. Kliewer, Regulation of CYP3A gene transcription by the PXR Annu. Rev. Pharmacol. Toxicol. 2002. 42:1-23].
Всего несколько десятилетий тому в печени крыс, подвергнутых воздействию фенилэтилбарбитуровой кислоты, был идентифицирован первый индуцибельный P450 (теперь классифицируемый как CYP2B) [Gonzalez и другие, 1993] и выдвинут постулат, подтвержденный только недавно, что посредником индукции является рецепторный белок. Позже был идентифицирован второй уникальный P450 (теперь классифицируемый как CYP1A), индуцируемый арилгидрокарбоном и 3-метилхолантреном, и детально описана его регуляция лиганд-активируемым рецептором. В начале 1980-ых, несколько групп исследователей идентифицировали третью форму P450 (теперь классифицируемую как CYP3A), индуцируемую веществами стероидной природы.
Эти исследования обеспечили понимание многих фармакологических, биохимических и биофизических свойств P450 ферментов, включая белковую структуру, специфику субстратов и кинетику фермента. В процессе развития молекулярной биологии стало возможным раскрытие особенностей структуры и функции гена P450, что объяснило феномены наблюдаемые фармакологами в более ранних исследованиях.
Среди цитохромов P450 печени и кишечника человека изоформа CYP3A является наиболее широко представленной составляя примерно 60 % от общего количества цитохромов в данных органах и около 30% - в организме. Она индуцируется многими субстратами, включая естественные и синтетические глюкокортикоиды (дексаметазон, тестостерон, кортизол) 6-beta-гидроксилирование которых она катализирует, прегнан-производные (прегненолон, 16-карбонитрил, прогестерон), антибиотики с макроциклическим лактонным кольцом (рифампицин, эритромицин), цитостатики и многие другие. Ферменты этой группы принимают участие в метаболизме примерно 60% всех лекарственных препаратов и отвечают за возникновение лекарственных взаимодействий практически во всех случаях, когда это касается индукции микросомальных ферментов или их ингибирования [Wrighton и Stevens, 1992; Gonzalez и другие., 1993; Cytochrome P450 enzyme regulation by induction and inhibition, Francois BERTHOU; Ross A McKinnon, Allan M Evans, 2000;].
Таблица 1 - Каталитическая активность к CYP3A субстратам в микросомах печени человека
| Субстраты | CYP изоформы экспрессирующиеся в клетках | Каталитическая активность к CYP3A субстратам в микросомах печени человека | |||||||||
| 3А4 | 3А5 | 3А7 | взрослые (%) | дети (%) | новорожденные(%) | зародыши (%) | ссылки | комменатрии | |||
| Лекарственные препараты | |||||||||||
| Карбамазепин | +++ | ++ | + | Представлен | CBZ-E формирование (?) | 34, 93 | Зависимые от времени изменения метаболитов формирование CBZ-E у мертворожденных | ||||
| Циклоспорин (M1 формирование) | +++ | +++ | 28 | ||||||||
| Циклоспорин (M1 7 формирование) | +++ | ND | 28 | 3A5 сформировано меньшее количество метаболитов | |||||||
| Декстрометорфан | 100 | 30 | 94, 95 | 3ММ формирование (CYP3A) | |||||||
| Диазепам(малый) | 100 | 140(3-12 мес.) | 15 (<24ч после рождения); 40-50 (1-7 день после рождения) | <5 | 96 | Скорость формирования темазепама (CYP3A зависимое) | |||||
| Эритромицин | +++ | ND/++++ | 32, 23 | ||||||||
| Этилморфин | + | ++ | 100 | 100 | 33, 68 | ||||||
| Индинавир | 100 | 67(6-11 лет) | 32 | 32, 91 | |||||||
| Лидокаин | ++ | +++ | ? | 97 | |||||||
| Мидазолам (формирование 1-гидрокси) | + | +++ | +/- | 5, 31 | |||||||
| Мидазолам (формирование 4-гидрокси) | ++ | +++ | 31,98 | ||||||||
| Нефидипин | +++ | +++ | 100 | 44 | 18 | 28, 32,90 | |||||
| Парацетамол (ацетаминофен) | 100 | 10 | 99 | CYP2E1, сульфатация и глюкуронидация | |||||||
| Квинидин | +++ | ND | 32 | ||||||||
| Зонисамид | +++ | + | ++ | 34 | |||||||
| Эндогенные субстраты | |||||||||||
| Андростанедион (6β-гидроксиляция) | +++ | ++ | ++ | 28,100 | |||||||
| Кортизол | +++ | ++ | - | 32,101 | |||||||
| DHEA (16a-гидроксиляция) | + | +++ | + | +++ | 5,32,37, 87 | ||||||
| DHEA (16β-гидроксиляция) | +++ | + | 87 | ||||||||
| DHEA-S | ++ | ++ | +++ | 32,34 | |||||||
| Прогестерон (6β -гидроксиляция) | +++ | ++ | 28,39 | ||||||||
| Тестостерон (2 β -гидроксиляция) | +++ | ++ | ++/++++ | <1 или 100 | 12-38 | 12 | 5,27,34, 90 | ||||
| Тестостерон (6 β -гидроксиляция) | +++ | ++ | + | 100 | 30-40 | 30-40 | 2-10 | 89,100, 102 | |||
| Тестостерон (15 β гидроксиляция) | +++ | +/- | 28 | ||||||||
| Ксенобиотики | |||||||||||
| Афлатоксин B1 | +++ | ND | 100 | 200 | 23,38,89, 103 | Активация афлатоксина B1 in vitro | |||||
| Бензпирена активация | Активность ? | +++ | 100 | 600 | 89 | ||||||
| Бензфетамин | +++ | +++ | 33,36 | ||||||||
| Гетероциклические амины | + | ++ | +++ | 104 | Активация IQ и MelQ от приготовленной мякоти, рыбы и табака | ||||||
| Стеригматоциклин | +++ | +++ | 100 | 50 | 38,89 | ||||||
Относительно взрослых = 100 %.
CBZ-E = карбамазепин-10,11-эпоксид;
DHEA = дегидроэпиандростерон;
DHEA-S = дегидроэпиандростерон 3-сульфат;
IQ = гетероциклический амин: 2-амино-3-диметил-имидазо[4.5-/]квинидин;
MelQ = гетероциклический амин :2-амино-3.4-диметил-имидазо[4,5-]квинидин;
3MM = 3-метокси-меторфан;
ND = не определяется; + к указаны увиличение уровней экспрессии; - нет указаных уровни экспресии; ? указанная экспрессия не известна.
Таблица 2 - Сравнительная характеристика основных CYP3A изоформ
| Изоформа Р-450 | Локализация | Ген | Процент от общего содержания | каталитическая активность, комментарии | Ссылки |
| CYP3A4 | мидзональные и центролобулярные регионы печени и энтероциты тонкой кишки (40 % от общего содержания), а так же CYP3A4 экспрессируется в пищеводе, двенадцатиперстной кишке, и ободочной кишке | 7 q22.1. Ген разделен на 13 экзонов и 12 интронов с длиной приблизительно 27КБ. | 30-40 | активность тестостерон p-гидроксилазы в микросомах печени человека, представляет главным образом активность CYP3A4, CYP3A4 также метаболизирует прокарциногены, например стеригматоцистин и афлатоксин В1. CYP3A активность немного выше у особей женского пола. Хотя менструальная фаза цикла не влияет на CYP3A4 активность. | 23,151, 48, 26, 32. |
| CYP3A5 | В печени- см. выше +CYP3A5, кажется, основная изоформа CYP3A экспрессирующаяся в желудке и пищеводе, а так же нисходящих отделах кишечника, почке, легких, крови (нейтрофилах и B лимфоцитах) и гипофизе (CYP3A5 mRNA и белок были обнаружены в человеческом гипофизе и расположены в клетках, продуцирующих гормон роста), а также незначительные количества в экстрагепатической эмбриональной и плодной ткани | На 83 %, гомологичен к CYP3A4 | 10-30 (до 74 %) в печени взрослых организмов | скорость образования 1-гирокси-мидазолама значительно выше с CYP3A5, формирование 4-гидрокси-мидазолама с CYP3A4 и CYP3A5 подобна, к эритромицину и этилморфину в 6 раз выше, чем у 3A4. Формирование карбамазепин-10,11 -эпоксида и 2-сулфамойлацетилфенола (SMAP) из зонисамида, катализируемого CYP3A5 составляет приблизительно 33 % и 10 %, по сравнению с CYP3A4. активно метаболизирует эстрадиол, дегидроэпиандростерон 3-сульфат и кортизол. | 56,62,63,64,6,15 34,68 311 13, 23 34 32, 33. |
| CYP3A7 | CYP3A7 является основной изоформой в печени эмбрионов, детей и новорожденных, хотя он присутствует и в печени взрослых но в меньшем колличестве, чем CYP3A4, CYP3A7 составляет приблизительно 32 % общего количества CYP содержание в плодной печени, незначительные количества в экстрагепатической эмбриональной и плодной ткани, Плацентарное и внутриматочное содержание CYP3A7, кажется, увеличивается знчительно от первого ко второму триместру беременности | Ген на 90% гомологичен 3А4 | 32 | Формирование 1-гидрокси-мидазолама и карбомазепина-10,11-эпоксида незначительно по сравнению с CYP3A4, Метаболизм зонисамида CYP3A7 составлял 70 % от уровня CYP3A4. биотрансформация цизаприда до норцизаприда или его метаболитов с 2 первичными гидроксилированными кольцамим CYP3A7гораздо (в 10 раз) выше, чем наблюдаемое у CYP3A4, CYP3A7 катализирует 16o-гидроксилирование дегидроэпиандростерон 3-сульфат, что является физиологически важной реакцией для формирования эстриола в период беременности, с более высокой афинитивностью и максимальной скоростью чем CYP3A4. CYP3A7 очень мало вовлечен в 6p-гидроксилирование тестостерона, CYP3A7 способен метаболизировать такие токсические вещества, как например вфлатоксин Bl | 18,19,36, 4,37 |
Множественные формы CYP3A присутствуют в печени крыс и мышей, одна изоформа обнаружена у кроликов, у человека были идентифицированы четыре CYP3A гена [Gonzalez и другие, 1993; Domanski и другие, 2001]. CYP3A гены кодируют наиболее распространенные CYP ферменты у людей, включая CYP3A4, CYP3A5, CYP3A7, и CYP3A43 [Wildt и другие, 1999; Gellner и другие, 2001]. Считается, что CYP3A4 является преобладающей формой в печени взрослого человека. CYP3A5 является полиморфной формой, чье присутствие в печени взрослого человека выражено переменной степенью. CYP3A5, как полагают, присутствует в печени, приблизительно у 20 % европейцев, однако последние исследования свидетельствуют о том, что, CYP3A5 экспрессируется и в печени более чем у 50 % негров [Lown и другие, 1994; Wildt и другие., 1999; Wandel и другие., 2000; Kuehl и другие., 2001] CYP3A4 и CYP3A5 распределены в множественных тканях, включая не только печень, но также кишечник и почки [Thummel и Wilkinson, 1998; Guengerich, 1999]. CYP3A7 - изоформа, найденная в тканях кишечника, репродуктивных органах, и печени младенцев, представлена также и в печени некоторых взрослых людей [Kitada и другие, 1985; Schuetz и другие., 1994]. Недавно был идентифицирован новый член семейства CYP3A (CYP3A43) - CYP3A43 mRNA найдена в основном в простате взрослого человека, хотя также представлены и в других тканях, включая печень, где индуцируется рифампицином [Gellner и другие, 2001] Westlind и другие. (2001) утверждают, что уровни CYP3A колеблются в печени на протяжении жизни индивидуума [Shimada и другие., 1994; Oesterheld, 1998]
Еще начальные фармакологические исследования продемонстрировали индукцию CYP3A глюкокортикоидами и, как это ни парадоксально, антиглоюкокортикоидами, что привело к идее существования неклассического глюкокортикоид-рецепторного механизма была и в итоге - к идентификации уникального представителя ядерного семейства рецепторов, прегнан X рецептора (PXR; NR1I2). Становилось все более и более очевидно, что PXR, также как другие ядерные рецепторы, посредник в процессе индукции CYP3A, где играет сложную роль, включая индуцибельность структурно разнообразными составами и обладает поразительными внутривидовыми различиями в индукционных профилях. Понимание значения роли ядерных рецепторов в регуляции экспрессии CYP3A и других генов P450 семейства предполагает дальнейшее определение физиологически функциональных и внутривидовых различий CYP3A в состоянии здоровья и болезни у человека.
Прегнан X рецептор (PXR)- представитель суперсемейства ядерных рецепторов, который появился в связи с развитием протекции организма от токсических химических веществ. PXR активируется структурно разнообразным спектром ксенобиотиков, включая широко используемые лекарственные препараты. Различные липофильные составы, продукты жизнедеятельности организма, такие как, желчные кислоты и стероиды, также активизируют PXR. PXR стимулирует транскрипцию цитохрома P450 3A монооксигеназ и других генов, вовлеченные в детоксикацию и элиминацию потенциально токсических веществ. Исследования PXR активации имеют важное значение для проектирования будущих лекарственных препаратов по двум причинам: с одной стороны, они могут использоваться, для определения возможностей отдельного лекарственного средства индуцировать экспрессию CYP3A гена и его взаимодействия с другими лекарственными препаратами. С другой стороны, PXR агонисты могут оказаться полезными в лечении заболеваний, при которых присходит аккумуляция токсических метаболитов, например холестатический синдром печени.
PXR включает стероид, ретиноид и рецепторы гормона щитовидной железы. Члены данного семейства функционируют как лиганд-активизируемые факторы транскрипции и играют основную роль в почти каждом аспекте развития индивидуума, а так же в физиологии взрослого организма. Члены семейства имеют общую структуру домена, которая включает высококонсервативную DNA, связанную с доменом (DBD) двумя “цинковыми пальцами”. DBD является рецептором-мишенью для коротких отрезков DNA (так называемых элементов ответа) в регуляторных областях целевых генов. Карбокси-терминалый регион ядерных рецепторов включает сохраненный лигандсвязующий домен (LBD), который служит сайтом состыковки лигандов, а также содержит мотивы димеризации и домены активации транскрипции (активатор функции - 2 (AF-2) геликса). Прикрепление лиганда к LBD заканчивается конформационным изменением в AF-2 геликсе, которое позволяет ядерному рецептору взаимодействовать с акцессорными белками и активизировать экспрессию целевых генов [48, 57, 58] [Bryan Goodwin1, Matthew R. Redinbo2, and Steven A. Kliewer , Regulation of CYP3A gene transcription by the PXR,Annu. Rev. Pharmacol. Toxicol. 2002. 42:1-23.].
Кульминацией вышеуказанных исследований можно считать признание того, что ядерные рецепторы подобные охарактеризованным для стероидных гормонов являются средствами индукции CYP3A. Таким образом, обширные данные, накопленные в процессе изучения этой группы физиологических лиганд-активируемых факторов транскрипции могут теперь быть расширены на всю систему метаболизма лекарственных средств.
Имеется большое количество факторов, способных воздействовать на токсичность и биотрансформацию лекарственных средств. Они обычно классифицируются на генетические и не-генетические, а также факторы окружающей среды. В последней категории, химическая экспозиция, в случае назначения лекарственных препаратов, профессиональные вредности на рабочем месте или загрязнение окружающей среды, воздуха и воды, ведет или к индукции или к ингибированию токсичности и метаболизма химических веществ.
Индукция определена как увеличение количества и активности метаболизирующего препарат фермента (DME), главным образом цитохромом P450 семейств от 1 до 4. Это длительное (часы и дни) последствие экспозиции химическим веществом.
Ингибирование ферментов P450 означает любое снижение уровня метаболизма специфического ксенобиотика другим ксенобиотиком, за счет одновременного присутствия в активном сайте фермента. В CYP3A семействе наблюдается также, активация P450 фермента аллокринными процессами.
Итак, классическое определение индукции - de novo синтез новых молекул фермента в результате увеличения транскрипции его гена после стимуляции соответствующим химическим сигналом.
Интересным можно считать сообщение о том, что гормон роста и трийодтиронин регулируют гидроксилирование стероидов цитохромом P-450 (CYP) 3A изоформой, которая конститутивно экспрессируются главным образом в перивенозных областях печени. Сравнивая лизаты клеток, полученные от перипортальных и перивенозных ацинарных областей наблюдалось увеличение CYP3A экспрессии после гипофизэктомии, что происходило главным образом благодаря увеличению экспрессии в обычно малоактивной перипортальной области. Этот эффект был особенно сильно выражен у особей женского пола. Прием гормона роста восстанавливал перивенозный уровень экспрессии, что было подтверждено иммуногистохимическим анализом секций печени. Анализ перипортальных и перивенозных mRNA обратно транскриптазной PCR демонстрирует, что у мужчин изменения CYP3A2 mRNA происходят паралельно с изменениями на белковом уровне. У женщин, CYP3A2 mRNA был обнаружен только после гипофизэктомии, поэтому казалось, что зональные белковые изменения предшествуют изменениям уровням CYP3A1 mRNA. Применение животными с гипофизэктомией трийодтиронина также подавляло экспрессию CYP3A, как у самцов, так и у самок.
Однако, это произходило почти исключительно в перипортальных областях, что определялось и на белковом уровне, методами иммуноблоттинга и иммуногистохимически, а так же на CYP3A1 и 3A2 mRNA уровнях. Эти результаты указывают на то, что гормон роста и гормон щитовидной железы регулируют экспрессию CYP3A генов зоно- специфически, подавляя их транскрипцию в перипортальных областях печени. [Oinonen T, Lindros KO. Hormonal regulation of the zonated expression of cytochrome P-450 3A in rat liver.Biochem. J. (1995) 309, (55–61) (Printed in Great Britain)] Известно, что большинство взаимодействий лекарственных средств, посредством CYP3A - результат или индукции или ингибирования этого фермента [241]. CYP3A4 активность может быть индуцирована in vitro и in vivo кортикостероидами (например дексаметазоном), противосудорожными средствами (например фенилэтилбарбитуровая кислота, фенитоин, карбамазепин) и некоторыми антибиотиками например рифампицин, рифапентин (рифампицин стимулирует 7-этоксикумарин O-деэтилирование (ECOD) активность в микросомах человеческих легкого. Однако многие известные индукторы CYP3A4, такие как дексаметазон, рифампицин или фенилэтилбарбитуровая кислота, не индуцируют CYP3A5 активность in vitro. Некоторые индукторы, однако, увеличивают в печени CYP3A белковые уровни у животных при помощи стабилизации. Такая посттранскрипционная регуляция сначала демонстрировалось для тролеандомицина и других антибиотиков с макроциклическим лактонным кольцом, которые образуют устойчивые комплексы с ферментом. Механизм, благодаря которому это взаимодействие стабилизирует CYP3A относительно последующей деградации, неясен, но может быть связано с процессом цАМФ-зависимого фосфорилирования , который принимает участие в белковой денатурации (159)[Thummel K.E., In vitro and in vivo drug interactions involving human CYP3A.,Annu. Rev. Pharmacol. Toxicol. 1998. 38:389-430]
В современных изучениях, использующих субстраты, связанные в активном гомологичном сайте CYP3A4 и моделированные подобно бактериальному CYPBM-3, было доказано, что водородная связь большинства субстратов с Asn74 остатком CYP3A4 происходит часто наряду с ![]() составляющей Phe72 [Lewis DFV, Eddershaw PJ, Goldfarb PS and Tarbit MH (1996) Molecular modelling of CYP3A4 from an alignment with CYP102: Identifcation of key interactions between putative active site residues and CYP3A- specific chemicals. Xenobiotica 10: 1067-1086.; Tomlinson ES, Lewis DFV, Maggs JL, Kroemer HK, Park BK and Back DJ (1997) In vitro metabolism of dexamethasone (DEX) in human liver and kidney: The involvement of CYP3A4 and CYP17 (17,20 LYASE) and molecular modelling studies. Biochem Pharmacol 54: 605-611; Lewis DFV and Lake BG (1998) Molecular modelling and quantitative structure-activity relationship studies on the interaction of omeprazole with cytochrome P450 isozymes. Toxicology 125: 31-44] . Точно так же водородная связь с Asn74, кажется, происходит с ингибиторами подобными кетоконазолу и гестодену [Lewis DFV, Eddershaw PJ, Goldfarb PS and Tarbit MH (1996) Molecular modelling of CYP3A4 from an alignment with CYP102: Identifcation of key interactions between putative active site residues and CYP3A- specific chemicals. Xenobiotica 10: 1067-1086.]. Кроме того, Льюис и др. [Lewis DFV, Eddershaw PJ, Goldfarb PS and Tarbit MH (1996) Molecular modelling of CYP3A4 from an alignment with CYP102: Identifcation of key interactions between putative active site residues and CYP3A- specific chemicals. Xenobiotica 10: 1067-1086.] утверждают, что предполагаемые структурные требования CYP3A4 субстратов включают водород-связующий акцепторный атом, который находится на расстоянии 5.5 до 7.8 Å от сайта метаболизма, который в свою очередь находится на 2 до 4 Å от молекулы кислорода в геме. Дальнейшая молекулярная модель CYP3A4 основанна на ее последовательном сравнении со всеми четырьмя бактериальным CYPS, и наводит на мысль, о взаимодействии множественных гидрофобных областей с субстратами прогестероном и эритромицином [Szklarz GD and Halpert JR (1997) Molecular modeling of cytochrome P4503A4. J Comp Aided Mol Des 11: 265-272.]. Дополнительные изучения, проведенные Halpert и др. [Harlow GR and Halpert JR (1997) Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P4503A4. J Biol Chem 272: 5396-5402; Domanski TL, Liu J, Harlow GR and Halpert JR (1998) Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: Role in steroid hydroxylase activity and alpha -napthoflavone stimulation. Arch Biochem Biophys 350: 223-232.] с использованием аланин-сканирующего мутагенеза идентифицировали остатки аминокислоты, необходимые для субстратной специфики и флавоноид активации CYP3A4. [Three-Dimensional-Quantitative Structure Activity Relationship Analysis of Cytochrome P-450 3A4 Substrates., Sean Ekins, Gianpaolo Bravi , James H. Wikel and Steven A. Wrighton]
составляющей Phe72 [Lewis DFV, Eddershaw PJ, Goldfarb PS and Tarbit MH (1996) Molecular modelling of CYP3A4 from an alignment with CYP102: Identifcation of key interactions between putative active site residues and CYP3A- specific chemicals. Xenobiotica 10: 1067-1086.; Tomlinson ES, Lewis DFV, Maggs JL, Kroemer HK, Park BK and Back DJ (1997) In vitro metabolism of dexamethasone (DEX) in human liver and kidney: The involvement of CYP3A4 and CYP17 (17,20 LYASE) and molecular modelling studies. Biochem Pharmacol 54: 605-611; Lewis DFV and Lake BG (1998) Molecular modelling and quantitative structure-activity relationship studies on the interaction of omeprazole with cytochrome P450 isozymes. Toxicology 125: 31-44] . Точно так же водородная связь с Asn74, кажется, происходит с ингибиторами подобными кетоконазолу и гестодену [Lewis DFV, Eddershaw PJ, Goldfarb PS and Tarbit MH (1996) Molecular modelling of CYP3A4 from an alignment with CYP102: Identifcation of key interactions between putative active site residues and CYP3A- specific chemicals. Xenobiotica 10: 1067-1086.]. Кроме того, Льюис и др. [Lewis DFV, Eddershaw PJ, Goldfarb PS and Tarbit MH (1996) Molecular modelling of CYP3A4 from an alignment with CYP102: Identifcation of key interactions between putative active site residues and CYP3A- specific chemicals. Xenobiotica 10: 1067-1086.] утверждают, что предполагаемые структурные требования CYP3A4 субстратов включают водород-связующий акцепторный атом, который находится на расстоянии 5.5 до 7.8 Å от сайта метаболизма, который в свою очередь находится на 2 до 4 Å от молекулы кислорода в геме. Дальнейшая молекулярная модель CYP3A4 основанна на ее последовательном сравнении со всеми четырьмя бактериальным CYPS, и наводит на мысль, о взаимодействии множественных гидрофобных областей с субстратами прогестероном и эритромицином [Szklarz GD and Halpert JR (1997) Molecular modeling of cytochrome P4503A4. J Comp Aided Mol Des 11: 265-272.]. Дополнительные изучения, проведенные Halpert и др. [Harlow GR and Halpert JR (1997) Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P4503A4. J Biol Chem 272: 5396-5402; Domanski TL, Liu J, Harlow GR and Halpert JR (1998) Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: Role in steroid hydroxylase activity and alpha -napthoflavone stimulation. Arch Biochem Biophys 350: 223-232.] с использованием аланин-сканирующего мутагенеза идентифицировали остатки аминокислоты, необходимые для субстратной специфики и флавоноид активации CYP3A4. [Three-Dimensional-Quantitative Structure Activity Relationship Analysis of Cytochrome P-450 3A4 Substrates., Sean Ekins, Gianpaolo Bravi , James H. Wikel and Steven A. Wrighton]
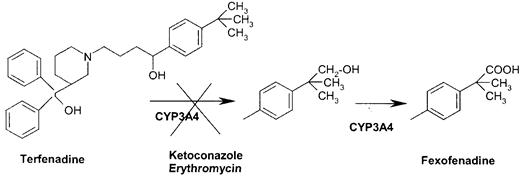
Необходимо отметить, что ингибирование фермента, метаболизирующего лекарственное средство, - возможно наиболее частая причина, лежащая в основе клинически важного фармакокинетического взаимодействия лекарственных средств. Ферменты цитохрома P450 (CYP) отвечают за большую часть 1 фазы метаболизма лекарственных средств. По этой причине, вопросы ингибирования этих ферментов - клинически важная проблема из за которой приходилось неоднократно удалять лекарственные препараты с рынка. Например, антигистаминный препарат терфенадин, который подвергается быстрому и интенсивному метаболизму путем гидроксилирования метильной группы, которая далее окисляется до активного метаболита фексофенадина, в обоих случаях основная роль принадлежит CYP3A4. (Рисунок 6.) При совместном приеме с ингибиторами CYP3A подобными кетоконазолу и эритромицину, происходит ингибироваение метаболизма препарата, ведя к серьезному увеличению концентрации терфенадина, стимулируя желудочковые аритмии.
Рис. 6 - Метаболизм терфенадина
Антидиабетическое лекарственное средство троглитазон было удалено с рынка лекарственных средств из-за инцидентов, связанных с возникновением явлений фатальной гепатотоксичности [N. Engl. J.Med 338, 916, 1998; Drug Metab. Dispo 27, 1260, 1999].
Как извесно, примерно 5% всех жителей США имеют генетический дефицит изоформ цитохрома 2-6, который участвует в метаболизме бета - блокаторов, нейролептиков и антидепрессантов. У этих пациентов не бывает ингибирования этой формы фермента хинидином, что наблюдается у всей остальной популяции. Ингибирование изоформы 3А встречается часто и вызывается большим количеством препаратов, часто применяющихся в практике. Это могут быть: кетоконазол, флюконазол, циклоспорин, ритонавир, дилтиазем, нифедипин, никардипин, флуоксетин, хинидин, верапамил и эритромицин (Таб.2). [Ross A McKinnon, 2000]
Таблица 2 - Ферменты цитохрома 450 3А вовлеченные в лекарственный метаболизм.
| Изоформа Р-450 | Вариабельность (Уровни) | Типичные индукторы | Специфические субстраты, реакции. | Селективные ингибиторы |
| CYP3A4/5/7 | 10-30 | Рифампицин (макролиды) Дексаметазон (кортикостероиды, прегненолон 16a-карбонитрил) Фенобарбитал; HIV ингибиторы протеаз, | Тестостерон 6-P-гидроксилирование, нифидипин дегидроксилирование, мидазолам 1-гидроксилирование, Циклоспорин (50% лек.средств), Толбутамид гидроксилирование, таксол 6a-гидроксилирование, Диклофенак гидроксилирование (S) Мефенетоин 4гидроксилирование | Кетоконазол, гестоден триацетилолеандомицин |
Антибиотики с макроциклическим лактонным кольцом могут стимулировать их собственную печеночную биотрансформацию до нитрозоалканов. Эти метаболиты получены при метаболическом окислении ±N(CH3)2 группы антибиотика и передаче ±NO группы. Нитрозоалканы впоследствии формируют инактивные CYP 3A4-iron-метаболит комплексы (Рисунок 8) результатом является ингибирование CYP 3A4-зависимой каталитической активности [3/ Pessayre D, Larrey D, Funck-Brentano C, et al.Drug interactions and hepatitis produced by some macrolideantibiotics. J Antimicrob Chemother 1985; 16(Suppl H): 181±194.]. Этот механизм объясняет большинство взаимодействий лекарственных средств, с участием антибиотиков с макроциклическим лактонным кольцом [4/ Periti P, Mazzei T, Mini E, Novelli A. Pharmacokinetic drug interactions of macrolides. Clin Pharmacokin 1992; 23:106±131.].
Вещества с макроциклическим лактонным кольцом отличаются по их способностям связаться и ингибировать изоформу CYP 3A4 цитохром P-450 [3/ Pessayre D, Larrey D, Funck-Brentano C, et al.Drug interactions and hepatitis produced by some macrolide antibiotics. J Antimicrob Chemother 1985; 16(Suppl H):181±194., 5/ Gascon PM, Dayer P. Comparative effects of macrolide antibiotics on liver mono-oxygenases. Abstract. Clin PharmacolTher 1991; 49: 158.7. 6/ Ohmori S, Ishii I, Kurina SI, et al. Effects of clarithromycin and its metabolites on the mixed function oxydase system in hepatic microsomes of rats. Drug Metab Dispos 1993; 21:358±363.,7/ Lindstrom TD, Hanssen BR, Wrighton SA. Cytochrome P-450 complex formation by dirithromycin and other macrolides in rat and in human liver. Antimicrob Agents Chemother 1993; 37: 265±269.]. Вышеуказанные различия побудили von Rosenstiel & Adam [8/ Von Rosenstiel NA, Adam D. Macrolide antibacterials. Drug interactions of clinical signiÆcance. Drug Safety 1995; 13: 105±122.] классифицировать макролиды на три группы на основе данных, обеспеченных в экспериментах in vitro.
Группа 1 включает эритромицин и тролеандомицин. Оба лекарственных препарата прочно связываются и ингибируют CYP 3A4.
Кларитромицин принадлежит ко 2 Группе. Его Эксгибиция имеет более низкую степень родства к CYP 3A4, по сравнению, с эритромицином, и формирование комплексов происходит в меньшей степени;
Группа 3 включает азитромицин и диритромицин. Эти составы продемонстрировали слабую степень взаимодействия с системой цитохрома P-450 in vitro.
Кларитромицин проявляет сходство с эритромицином в некоторых взаимодействиях лекарственных средств (например с психотропными средствами) что основано на результатах клинических исследований. Кроме того, множество данных, полученных при анализе клинических случаев демонстрируют, что азитромицин и диритромицин имеют потенциал для взаимодействий лекарственных средств, хотя гораздо меньшей степени чем эритромицин. Несоответствие между тем, что ожидается от данных, полученных in vitro и тем, что может наблюдаться в клинической практике, подчеркивает известную внутривидовую вариабильность степени каталитической активности цитохрома P-450 (от 10 до 20 уровней)
Не вызывает сомнений, что гетерогенность способностей пациентов метаболизировать посредством P-450 некоторые лекарственные препараты является в значительной степени результатом генетических [1/ Watkins PB. Drug metabolism by cytochrome P-450 in the liver and small bowel. Gastroenterol Clin N Am 1992; 21: 511±526.] факторов. Кроме лекарственной индукции/ингибирования ферментативных процессов, некоторые негенетические факторы вероятно вносят вклад во внутривидовые различия содержания в печени или активности П-450а:
-диетические факторы могут вносить достаточно существенный вклад.
-3A4 активность не может быть специфически угнетена заболеваниями, например циррозом печени [11/ Westphal JF, Brogard JM. Clinical pharmacokinetics of newer
antibacterial agents in liver disease. Clin Pharmacokin 1993; 24: 46±58.] или патологиями, связанными с желчевыводящей системой.
-Значительное влияние могут оказывать инфекционные процессы на активность CYP; вирусные или бактериальные поражения легких демонстрировали снижение активности CYP. В последовательном изучении, проводимом на 14 пациентах с гипертермическим симптомом и клинически диагносцированной пневмонией, антипириновый клиренс в среднем был снижен на 36 %.
Поскольку метаболиты формируют, по крайней мере, 6 изоформ цитохрома P-450 печени, а именно CYP 1A2, CYP 2B6, CYP 2C8, CYP 2C9, CYP 2C18 и CYP 3A4 [14/ Engel G, Hofmann U, Heidemann H, et al. Antipyrine as a probe for human oxidative drug metabolism: identiÆcation of the cytochrome P 450 enzymes catalizing 4-hydroxy-antipyrine, 3-hydroxymethylantipyrine, and norantipyrine formation. Clin Pharmacol Ther 1996; 59: 613±623.]. Следовательно, инфекция, вероятно, стимулирует глобальное ингибирование каталитической активности цитохрома P-450. Это происходит вероятно из-за высвобождения эндотоксина или интерферона во время инфекционного процесса [15/ Williams SJ, Baird-Lambert JA, Farrell GC. Inhibition of theophyllin metabolism by interferon. Lancet 1987; ii: 939±941. , 16/ Kitaichi K, Takagi K, Ixase M, et al. Decreased antipyrine clearance following endotoxin administration: in vivo evidence of the role of nitric oxide. Antimicrob Agents Chemother 1999; 43: 2697±2701.]. Очевидно, что комбинация интерферон - или эндотоксин связанная депрессия CYP, и индуцированное макролидами ингибирование CYP 3A4 изоформы могла бы дать в результате увеличение метаболического взаимодействия. Дополнительный фактор, который может вносить вклад в индивидуальную вариабильность степени взаимодействий лекарственных средств, есть вовлечение P-гликопротеида (Pgp) в процесс фармакокинетики. Вариации экспрессии этого белка, индуцированные лекарственными средствами могут накладываться на изменения на уровне CYP каталитической активности. P-gp-ATP-зависимый трансмембранный насос, который конститутивно экспрессируется в нормальных тканях, включая желудочно-кишечный эпителий, каналикулярную мембрану печени, почечные и капиллярные эндотелиальные клетки в центральной нервной системы [17/ Thiebaut F, Tsuruo T, Hamada H, et al. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein. J Histochem Cytochem 1989; 37: 159±164. , 18/ Thiebaut F, Tsuruo T, Hamada H, et al. Cellular localization of the multidrug resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA 1987; 84: 7735±7738.]. P-gp, кажется, играет ключевую роль в абсорпции, распределении и элиминации многих антитуморных средств также как других лекарственных средств, например дигоксина или циклоспорина [19/ Tanigawara Y, Okamura N, Hirai M, et al. Transport of digoxine by human P-glycoprotein expressed in a porcine kidney epithelial cell line (LLC-PK1). J Pharmacol Exp Ther 1992; 263: 840±845., 20/ Saeki T, Ueda K, Tanigawara Y, et al. Human P-glycoprotein transports cyclosporin A and FK 506. J Biol Chem 1993; 268: 6077±6080.]. Необходимо отметить, что многие P-gp ингибиторы - также являются ингибиторами CYP 3A [21/ Kim RB, Wandel C, Leake B, et al. Interrelationship between substrates and inhibitors of human CYP 3A and P-glycoprotein. Pharm Res 1999; 16: 408±413.]. Однако, Wandel и др. [22] продемонстрировали, что не имеется значительной корреляции между способностями веществ, ингибирующих P-gp ингибировать CYP 3A. В случае с макролидами, эритромицин выявил реверсивные изменения в высокой мультилекарственной резистентности клеток, которые сохраняют более высокий уровень экспрессии P-gp, чем у чувствительных к лекарственному средству родительских клеток, [23/ Hofsli E, Nissen-Meyer J. Reversal of drug resistance by erythromycin: erythromycin increases the accumulation of actinomycin D and doxorubicin in multi-drug resistant cells. Int J Cancer 1989; 44: 149±154.], что объясняется подавляющим эффектом макролидов на P-gp экспрессию. Кларитромицин так же демонстрирует способность к снижению почечного клиренса дигоксина посредством ингибирования P-gp в почечных эпителиоцитах [24/ Wakasugi H, Yano I, Ito T, et al. Effect of clarithromycin on renal excretion of digoxin: interaction with P-glycoprotein. Clin Pharmacol Ther 1998; 64: 123±128.]. Учитывая важность проблемы взаимодействий лекарственных средств в клинике, воздействие экзогенных зимических соединений, считаем необходимым рассмотреть подробнее особенности их метаболизма.
Бензодиазепины.
Бензодиазепины алпразолам, мидазолам, темазепам, и триазолам являются известными субстратами CYP 3A4 [26/ Von Moltke LL, Greenblatt DJ, Schmider J, Harmatz JS, Shader RI. Metabolism of drugs by P450, 3A isoforms. Implications for drug interactions in psychopharmacology. Clin Pharmacokinet 1995; 29(Suppl 1): 33±44.].
1) Триазолам: Greenblatt и др. [27/ Greenblatt DJ, Von Moltke LL, Harmatz JS, et al. Inhibition of triazolam clearance by macrolide antimicrobial agents: in vitro correlates and dynamic consequences. Clin Pharmacol Ther 1998; 64: 278±285.] продемонстрировали, используя in vitro препарированные микросомы человеческой печени, что эритромицин и кларитромицин являются мощными ингибиторами, с подобными значениями IC50, процессов биотрансформации триазолама. В отличии от них, азитромицин оказался слабым ингибитором. Данные, полученные от клинического изучения, проводимого теми же авторами на здоровых добровольцах [27/ Greenblatt DJ, Von Moltke LL, Harmatz JS, et al. Inhibition of triazolam clearance by macrolide antimicrobial agents: in vitro correlates and dynamic consequences. Clin Pharmacol Ther 1998; 64: 278±285.]свидетельствуют о том, и эритромицин и кларитромицин значительно увеличивали пиковую плазменную концентрацию триазолама и область под сывороточной концентрационно-разовой кривой (AUC), длительность периода полураспада, и заметно снижали оральный клиренс этого бензодиазепина до 33 % и 22 % от исходных значений. Ингибирующий эффект кларитромицина был больше, чем таковой у эритромицина, что особенно заметно при анализе данных о периоде полураспада и AUC триазолама. Все фармакокинетические эффекты триазолама были увеличены путем совмесного приема с эритромицином и кларитромицином. Динамические взаимодействия совместимы с увеличением плазменных концентраций триазолама. Наиболее выраженные изменеия, как оценено электроэнцефалограммой и другими исследованиями, были связаны с комбинацией триазолам-кларитромицин. Напротив, азитромицин не произвел никакой эффект на кинетику или динамику триазолама. Следовательно, вышеуказанные исследования демонстрирует, что кларитромицин - по крайней мере столь же мощный ингибитор CYP 3A4 как эритромицин.
2) Алпазолам - другой бензодиазепин, на метаболический клиренс которого оказывает депрессирующее действие эритромицин. На здоровых добровольцах, принимающих эритромицин в дозе (400 мг) три раза в день, ежедневно в течение 10 дней наблюдали увеличение 62 % AUC (0,48 ч), уменьшение на 60 % орального клиренса и более чем двойное увеличение периода полураспада элиминации алпазолама, в случае назначения перорально, как монопрепарата [31/ YasuiNY,Otani K, Kaneko S, et al.Akinetic and dynamic study of oral alprazolam with and without erythromycin in humans: in vivo evidence for the involvement of CYP 3A4 in alprazolam metabolism. Clin Pharmacol Ther 1996; 59: 514±519.]. Резюмируя все вышесказанное о бензодиазепинах, можно отметить, что совмесное применение антибиотика с макроциклическим лактонным кольцом (за исключением азитромицина) с одним из бензодиазепинов имеющим высокую степень метаболизма, в клинических условиях нужно избегать, либо дозу бензодиазепинов существенно корегировать.
| Субстрат | Взаимодействующий макролид | Субьекты исследования | Режим введения, дозы. | Результаты | Действие | |
| Алпразолам | Эритромицин (31) | Здоровые добровольцы отобранные методом случайной выборки. | 400 мг.*10 дней | Увеличение на 62 % AUC, без изменений психомотроных фнукций(алпразолам применялся в виде монодозы орально) | Контрольные эффекты | |
| мидазолам | Эритромицин (28) Кларитромицин (29) Кларитромицин (30) Азитромицин (30) | Здоровые добровольцы отобранные методом случайной выборки. Здоровые добровольцы Последователь-но Здоровые добровольцы отобранные методом случайной выборки. | 500 мг*6 дней 500 мг.*7 дней 250 мг.*5 дней 500 мг*3 дня | Четырехкратное увеличение AUC, значительные изменения психомоторных функций Увеличение с 2.4 сгибами в оральной доступности мидазолама Увеличение с 3.5 сгибами AUC, усиление фармакодинамики без значительных фармакокинетических или динамических изменений | Избегать комбинации с эритромицином или кларитромицином, или коррегировать дозу мидазолама на 75 % | |
| триазолам | Кларитромицин (27) Кларитромицин (27) Азитромицин (27) | Здоровые добровольцы отобранные методом случайной выборки. | 500 мг*2 дня 500 мг*2 дня 500 мг*1 день, 250-2 день | Тройное увеличение AUC, усиление контролируемых динамических эффектов пятикратное увеличение AUC, усиление эффектов Никакое кинетическое или динамическое изменение не обнаружено. | Контрольные эффекты | |
| клозапин | Эритромицин (32) Эритромицин (32) | история болезни история болезни | 333 мг*3 дня 250 мг*7 дней | Увеличение сыворотчной концентрации клозапина, лейкоцитоз, сонливость, дезориентировка Двойное увеличение в сыворотке клозапина приступ тонико - клонических судорог | Избегать комбинации или уменьшать в два раза дозу клозапина и наблюдать за побочными эффектами | |
| пимозид | Кларитромицин (35) | Здоровые добровольцы отобранные методом случайной выборки. | 500 мг*5 дней | 113 % увеличение в AUC значительное увеличение интервала QT | Комбинация противопоказанна | |
| карбамазепин | Кларитромицин (25) Азитромицин (25) | Здоровые добровольцы отобранные методом случайной выборки. Здоровые добровольцы | 500 мг*5 дней 500 мг*3 дня | значительное уменьшение в AUC и Cmax метаболита эпоксида карбамазепина Без кинетических изменений | Снижение дозы карбамазепина на 25 %-50 % | |
| дизопирамид | Кларитромицин (52) | история болезни | 250 мг*6 дней | Желудочная фибрилляция отмечена увиличением QT (625 м.с) | наблюдение за ЭКГ и плазменной концентрацией лекарственного средства. | |
| квинидин | Эритромицин (49) | Здоровые добровольцы Последователь-но | 250 мг*7 дней | уменьшение на 34 % общего клиренса Cmax увеличенно на 39 % | Контроль ЭКГ, сывороточной концентрации и факторов, предраспологающих к трепетанию - мерцанию миокарда | |
| цизаприд | Кларитромицин (40) Кларитромицин (41) | история болезни Здоровые добровольцы отобранные методом случайной выборки. | 500 мг*3 дня 500 мг*5 дней | Полиморфная желудочная тахикардия QT интервал увеличился до 640 м.с. Тройное увеличение AUC увеличение интервала QT 25м.с. | Комбинация противопоказанна | |
| симвастатин | Эритромицин (45) | Здоровые добровольцы отобранные методом случайной выборки. | 500 мг*2 дня | увеличение в AUC c 6.2 сгибами. | Избегать комбинации, или контролировать сывороточную креатин киназу. и наличие запаха Сельдерея от мышц. | |
Нейролептические средства.
1) Клозапин - новое нейролептическое средство, используемое при лечении шизофрении, стойкой к другим нейролептикам. Эритромицин взаимодействовал с этим средством, посредством ингибирования его метаболического клиренса, что выражалось в серьезных побочных эффектах включая сонливость, дезориентировку, расстройства координации движений и способности передвигаться, связанные с совместным применением эритромицина и клозапина (Таблица 3) [32/ Glassner Cohen LG, Chesley S, Eugenio S, et al. Erythromycin-induced clozapin toxic reaction. Arch Intern Med 1996; 156: 675±677., 33/ Funderberg LG, Vertrees JE, True JE. Seizure following addition of erythromycin to clozapin treatment. Am J Psychiatry1994; 151: 1840±1841.]. В фармакокинетическом исследовании, кларитромицин в дозировке (500 мг) два раза в день, ежедневно, в течение 5 дней, ингибировал метаболический клиренс (CYP3A-зависимый) другого нецролептика -пимозида, в результате получены данные о повышении плазменной концентрации, значительном увиличении интервала QT и увиличении риска развития явлений кардиотоксичности [35/ Desta Z, Kerbusch T, Flockhart DA. Effect of clarithromycin on the pharmacokinetics and pharmacodynamics of pimozide in healthy poor and extensive metabolizers of cytochrome P-450 2D6 (CYP 2D6). Clin Pharmacol Ther 1999; 65: 10±20.].
Цизаприд.
Цизаприд- широко используемое лекарственное средство при желудочно-пищеводном рефлюксе, гастропарезе, и диспепсии. Этот препарат подвергается интенсивному первичному метаболизму, и в печени и кишечнике [36/ Barone JA, Jessen LM, Colaizzi JL, Bierman RH. Cisapride: a gastrointestinal prokinetic drug. Ann Pharmacother 1994; 28:488±500.Macrolide-induced metabolic drug interactions
f2000 Blackwell Science Ltd Br J Clin Pharmacol, 50, 285±295 293]. Случаи возникновения тахикардии и экстрасистолии при приеме цизаприда известны и число их уже более чем 13000[37/ Imman W, Kubota K. Tachycardia during cisapride treatment.Br Med J 1992; 305: 1019.]. Первые сообщения о возникновении аритмии, как результата взаимодействия между лекарственными препаратами было получено при совместном применении цизаприда с эритромицином [38/ Bran S, Murray W, Hirsch IB, Plamer JP. Long QT syndrome during high-dose cisapride. Arch Intern Med 1995; 155:765±768.]; наблюдалось увеличение интервала QT на 550м.с. от нормального начального значения с прогрессией направленной к полиморфной неподтвержденной желудочной тахикардии. Более чем 50 % сообщений о трепетании, мерцании миокарда, удлиннении интервалов QT, и смертных случаях, связанных с приемом цизаприда связаны с его взаимодействием с препаратами, известными, как ингибиторы CYP3A4. [39/ Wysowski DK, Bacsanyi J. Cisapride and fatal arrhythmia. NEngl J Med 1996; 335: 290±291.]. Факторы риска для развития аритмии были идентифицированы, ими оказались наличие в анамнезе стенокардии и аритмий, почечной недостаточности, и нарушений электролитного баланса (включая гипокалиемию, гипомагниемию, и гипокальциемия) [39/ Wysowski DK, Bacsanyi J. Cisapride and fatal arrhythmia. NEngl J Med 1996; 335: 290±291.]. Кларитромицин демонстрирует вышеуказанные побочные явления при назначении в комбинации с цизапридом [40/ Piquette RK. Torsade de pointes induced by cisapride/clarythromycin interaction. Ann Pharmacother 1999;33: 22±26]. Данные полученные при анализе историй болезни и независимых исследований относительно этого взаимодействия представлены в Таблице 1. Потенцирование кардиотоксического эффекта цизаприда, следующего из ингибирования CYP 3A4 ± связанного метаболизма производится при совместном приеме с кларитромицином [40/ Piquette RK. Torsade de pointes induced by cisapride/clarythromycin interaction. Ann Pharmacother 1999; 33: 22±26. 41/ Van Haarst AD, Van Kloster GA, Van Gerven JM, et al. The inØuence of cisapride and clarithromycin on QT intervals in healthy volunteers. Clin Pharmacol Ther 1998; 64: 542±546.] – что является механизмом, лежащим в основе побочной реакции лекарственного препарата.
Ингибиторы HMG-CoA редуктазы.
Эти средства метаболизируются CYP 3A4 и оказывают связанные с дозой токсические эффекты на скелетную мышцу, начиная с рассеянной миалгии и миопатии вплоть до острого рабдомиолизиса [44/ Illingworth DR, Tobert JA. A review of clinical trials comparing HMG-CoA inhibitors. Clin Ther 1994; 16:366±385.]. В клиническом фармакокинетическом исследовании, эритромицин в дозе (500 мг) два раза в сутки, ежедневно, в течение 2 дней увеличивал AUC симвастатина в сыворотке в шесть раз [45/ Kantola T, Kivisto KT, Neuvonen PJ. Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther 1998; 64: 177±182.], что стало возможно именно благодаря игибирующему эффекту эритромицина на CYP 3A4. Для того что бы избежать таких осложнений, как острый рабдомиолизис необходимо избегать совместного применения эритромицина и симвастатина, а в случае с ловастатином ограничить суточную дозу до 20 мг [8/ Von Rosenstiel NA, Adam D. Macrolide antibacterials. Drug interactions of clinical signiÆcance. Drug Safety 1995; 13: 105±122.]. Рабдомиолизис также был в числе осложнений при назначении ловастатина и кларитромицина или азитромицина (одна история болезни каждого случая, Таблица 1) [46/ Grunden JW, Fisher KA. Lovastatin-induced rhabdomyolysis possibly associated with clarithromycin and azithromycin. AnnPharmacother 1997; 31: 859±863.]. Поэтому, неблагоприятные эффекты, подобно увиличения сывороточной креатин-киназы и мышечная слабость, должны насторожить при назначении одной из вышеупомянутых схем.
Антиаритмические средства.
Антиаритмическое средство 4 класса IA квинидин элиминируется из организма прежде всего посредством биотрансформации в печени, и приблизительно 50 % его метаболизма катализируется CYP 3A4 [47/ Guengerich FP, Muller Enoch D, Blair IA. Oxidation of quinidine by human liver cytochrome. Mol Pharmacol 1986; 30: 287±295.
]. Spinler и др. [48/ Spinler SA, Cheng JW, Kindwall KE, Charland SL. Possible
inhibition of hepatic metabolism of quinidine by erythromycin. Clin Pharmacol Ther 1995; 57: 89±94.] сообщили о том, что назначение внутривенно эритромицина лактобионата в дозе (1 г.) четыре раза в сутки, ежедневно, при длительной терапии квинидином закончилось уменьшением до 50 % общего клиренса квинидина после 5 дней терапии. В открытом клиническом исследовании на здоровых добровольцах, фармакокинетические аспекты применения разовой оральной дозы квинидина (200 мг), оценивали до и в течении терапии эритромицином в дозе (250 мг) 4 раза в сутки, в течении 7 дней [49/ Damkier P, Hansen LL, Brosen K. Effect of diclofenac,
disulÆram, itraconazole, grapefruit juice, and erythromycin on the pharmacokinetics of quinidine. Br J Clin Pharmacol 1999; 48: 829±838.]. Прием эритромицина снижал общий клиренс квинидина, а так же его частичный клиренс с 3 -гидроксилированием и N-окислением на 34, 50 и 33 %, среднее Cmax увеличевалось на 39 %, ингибирование актиности печеночного и кишечного CYP 3A4, за счет эритромицина, объясняло эти наблюдения, в то время как роль P-gp в этом процессе была вспомогательной. Были зарегистрированы случаи угрожающей жизни желудочковой аритмии, котроя являлась следствием взаимодействия между дизопирамидом и эритромицином [51/ Ragosta M, Weihl AC, Rosenfeld LE. Potentially fatal interaction between erythromycin and disopyramide. Am J Med 1989; 86: 465±466.].А один случай, о результатах взаимодействия с кларитромицином (Таблица 1) стал известен недавно [52/ Paar D, Terjung B, Sauerbruch T. Life±threatening interaction between clarithromycin and disopyramide. Lancet 1997; 249: 326±327.]. Во всех этих случаях, серьезная желудочковая аритмия была вызвана увиличением интервала QT, до 600 м.с.
Варфарин.
Имеются сообщения об увеличении гипопротромбинэмического эффекта варфарина натрия после назначения эритромицина [8/ Von Rosenstiel NA, Adam D. Macrolide antibacterials. Drug interactions of clinical signiÆcance. Drug Safety 1995; 13: 105±122., 25/ Amsden GW. Macrolides versus azalides: a drug interaction update. Ann Pharmacother 1995; 29: 906±917.]. Протромбиновое время увеличилось вдвое после 7 дней терапии и его увиличение связывались с возникающими осложнениями. Однако, имеется несоответствие между такими данными и изменениями, наблюдаемыми в фармакокинетических исследованиях [53/ Bachmann K, Schwartz JL, Forney R, Frogameni A, Jauregi LE. The effect of erythromycin on the disposition kinetics of warfarin. Pharmacology 1984; 28: 171±176. , 54/ Weibert RT, Lorentz SM, Townsend RJ, Cook CE, Klauber MR, Jagger PI. Effects of erythromycin in patients receiving long-term warfarin therapy. Clin Pharm 1989; 8: 210±214.]. Например в исследовании, проведенном Bachman и др. [53/ Bachmann K, Schwartz JL, Forney R, Frogameni A, Jauregi LE. The effect of erythromycin on the disposition kinetics of warfarin. Pharmacology 1984; 28: 171±176.], эритромицин снижал клиренс варфарина на 14 % у здоровых добровольцев. Варфарин - рацемическая смесь R- и S-варфарина, с S-формой связывают увеличение мощности препарата. Обе формы превращаются при метаболизме цитохромом P-450 с преобладающей причастностью CYP 1A1, CYP 1A2, CYP 2C9, CYP 2C19, и CYP 3A4 [55/ Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther 1997; 73: 67±74.]. S-варфарин превращается при обмене веществ прежде всего CYP 2C9, в то время как CYP 1A2 и CYP 3A4 преобладают в метаболизме R-варфарина. Относительно ограниченные изменения в фармакокинетике варфарина у здоровых добровольцев проявляются при назначении с эритромицином, когда происходит совместное ингибирование CYP 3A4, и в меньшей степени CYP 1A2 [2/ Slaughter RL, Edwards DJ. Recent advances: the cytochrome P ± 450 enzymes. Ann Pharmacother 1995; 29: 619±624. Jean Frederic Westphal 292 f2000 Blackwell Science Ltd Br J Clin Pharmacol, 50, 285±295, 56/ Chang TK, Gonzales FJ, Waxman DJ. Evaluation of triacetyloleandomycin, a-naphtho-Øavone and diethyldithiocarbamate as selective chemical probes for inhibition of human cytochrome P450. Arch Biochem Biophys 1994; 311: 437±442.]. Следовательно, это взаимодействие лекарственных средств будет увеличивать силу и продолжительность эффектов друг друга. Что же касается полусинтетических макролидов, то известно, что только четыре случая взаимодействия с варфарином были опубликованы, включая кларитромицин и азитромицин [59/ Recker MW, Kier KL. Potential interaction between clarithromycin and warfarin. Ann Pharmacother 1997; 31: 996±998.,60/ Gooderham MJ, Bolli P, Fernandez PG. Concomitant digoxin toxicity and warfarin interaction in a patient receiving clarithromycin. Ann Pharmacother 1999; 33: 796±799.,61/ Lane G. Increased hypoprothrombinemic effect of warfarin possibly induced by azithro-mycin (letter). Ann Pharmacother 1996; 30: 884±885., 62/ Woldtvedt BR, Cahoon CL, Bradley LA, Miller SJ. Possible increased nticoagulation effect of warfarin induced by azithromycin (letter). Ann Pharmacother 1998; 32: 269±270.]. Диритромицин не имеет никакого эффекта на протромбиновое ремя у здоровых добровольцев, получающих варфарин [42/ Watkins VS, Polk RE, Stotka JL. Drug interactions of macrolides: emphasis on dirithro-mycin. Ann Pharmacother 1997; 31: 349±356.] (Таблица 1).
Иммуносупрессоры.
Иммуносупрессор циклоспорин интенсивно метаболизируется CYP 3A , что само по себе создает значительный потенциал для взаимодействия с другими субстратами, [63/ Yee GC, McGuire TR. Pharmacokinetic drug interactions with cyclosporine (Part I). Clin Pharmacokinet 1990; 19: 319±332.]. Циклоспорин имеет низкий терапевтический индекс, и его почечная токсичность является связанной с его концентрацией. В сообщениях о многочисленных клинических исследованиях присутствуют данные о значительном увеличении AUC на фоне снижения клиренса циклоспорина после приема эритромицина [63/ Yee GC, McGuire TR. Pharmacokinetic drug interactions with cyclosporine (Part I). Clin Pharmacokinet 1990; 19:319±332.].
Считается, что не только CYP 3A4, но и Pgp играет роль в фармакокинетике циклоспорина. Gupta и др. [68/ Gupta SK, Bakran A, Johnson RW, Rowland M.
Erythromycin enhances the absorption of cyclosporin (letter). Br J Clin Pharmacol 1988; 25: 401±402.] считают, что эритромицин увеличивает абсолютную биодоступность перорально назначаемого циклоспорина. Этот эффект мог бы быть приписан снижению пресистемного метаболизма циклоспорина в кишечнике в результате эритромицин –индуцированного ингибирования CYP 3A4 кишечника. Однако, учитывая, что P-gp - также присутствует в кишечных эпителиоцитах [17/ Thiebaut F, Tsuruo T, Hamada H, et al. Cellular localization of the multidrug resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA 1987; 84: 7735±7738., 69/ Greiner B, Eichelbaum R, Fritz P, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin.J Clin Invest 1999; 104: 147±153.], и что эритромицин демонстрирует ингибирование экспрессии P-gp на линиях опухолевых клеток in vitro [23/ Hofsli E, Nissen-Meyer J. Reversal of drug resistance by erythromycin: erythromycin increases the accumulation of actinomycin D and doxorubicin in multi-drug resistant cells. Int J Cancer 1989; 44: 149±154.], увеличение биодоступности циклоспорина, при назачении его с эритромицином можно объяснить ингибированием CYP и P-gp и в печени и в кишечнике. Данные о взаимодействии между кларитромицином и циклоспорином с последующим увиличением токсичности циклоспорина были сообщены некоторыми авторами[8/ Von Rosenstiel NA, Adam D. Macrolide antibacterials. Drug interactions of clinical signiÆcance. Drug Safety 1995; 13: 105±122., 70/ Spicer ST, Liddle C, Chapman JR, et al. The mechanism of cyclosporin toxicity induced by clarythromycin. Br J Clin
Pharmacol 1997; 43: 194±196.]. Согласно Spicer и др. [70] (Таблица 1) основной механизм – индуцируемое макролидами ингибирование CYP 3A4, что ведет к снижению клиренса и увеличению концентрации в крови циклоспорина. Методом эксперимента, авторами изучалась особенности фармакокинетии циклоспорина при совместном применении с диритромицином на 15 стабильных почечных [71/ Bachmann K, Sullivan TJ, Reese JH, et al. The inØuence of dirithromycin on the pharma-cokinetics of cyclosporine in healthy subjects and in renal transplant patients. Am J Ther
1995; 2: 490±498.] трансплантах пациентов. Применение диритромицина в дозе (500 мг) ежедневно в течение 14 дней закончилось снижением на 17 % клиренса циклоспорина, и увеличеним на 16 % средней стационарной концентрации циклоспорина, увеличением на 13 % в стационарной концентрации циклоспорина. Резюмируя все, вышеизложенное можно сказать, что назначение макролидов пациентам в клинике с циклоспорином, требует постоянного контроля сывороточной концентрации циклоспорина и креатинина, для того чтобы позволить соответствующее регулирование дозировки циклоспорина.
Теофиллин.
Взаимодействия макрролидов с теофиллином хорошо изучены. В большинстве исследований, эритромицин и кларитромицин снижали клиренс теофиллина на 20±25 % после 7 дней совместной терапии [25/ Amsden GW. Macrolides versus azalides: a drug interaction update. Ann Pharmacother 1995; 29: 906±917.]. Это взаимодействие наиболее вероятно, в случае назначения относительно высоких доз (> 1.5 г. в день), и при длительной терапи эритромицином [74/ Prince RA, Wing DS, Weinberger MM et al. Effect of erythromycin on theophylline kinetics. J Allergy Clin Immunol 1981; 68: 427±431.]. Теофиллин метаболизируется у человека путем N-деметилирования и 8- гидроксилирования. Ингибиторы CYP 3A4 (включая тролеандомицин) ингибируют и N-деметилирование [56/56 Chang TK, Gonzales FJ, Waxman DJ. Evaluation of triacetyloleandomycin, a-naphtho-Øavone and diethyldithiocarbamate as selective chemical probes for inhibition of human cytochrome P450. Arch Biochem Biophys 1994; 311: 437±442.] и 8- гидроксилирование in vitro [77/ Tjia JF, Colbert J, Back DJ. Theophylline metabolism in human liver microsomes: inhibitory studies. J Pharmacol Exp Ther 1996; 276: 912±917.]. Однако, эти эксперименты показали, что CYP 3A4 ингибиторы снижали N-деметилирование максимумом на 16 %. На этом основании, хорошо изученное взаимодействие антибиотиков макролидов с теофиллином in vivo можно было бы объяснять ингибированием CYP 1A2 и CYP 3A4. Однако, учитывая относительно слабый ингибирующий эффект макролидов на CYP 1A2 активность in vitro, ингибирующий эффект макролидов на метаболизм теофиллина может быть усилен у субъектов демонстрирующих низкую активность CYP 1A2 и высокую активностьCYP 3A4, как последнего изофермента принимающего участие в метаболизме теофиллина [1/ Watkins PB. Drug metabolism by cytochrome P-450 in the liver and small bowel. Gastroenterol Clin N Am 1992; 21: 511±526.]. Эта гипотеза была обоснована недавно в многочисленных исследованиях in vitro [77/ Tjia JF, Colbert J, Back DJ. Theophylline metabolism in human liver microsomes: inhibitory studies. J Pharmacol Exp
Ther 1996; 276: 912±917.]. Такая гипотеза могла бы объяснить, почему множество предполагаемых клинических испытаний будет не в состоянии показывать статистически достоверное снижение клиренса теофиллина, при совместной терапии с эритромицином [4/ Periti P, Mazzei T, Mini E, Novelli A. Pharmacokinetic drug interactions of macrolides. Clin Pharmacokin 1992; 23: 106±131.].
Карбамазепин.
Данные из многочисленных историй болезни и некоторых исследований хорошо освещают взаимодействие между эритромицином и карбамазепином. Назначение карбамазепина с макролидом приводило к четырехкратному увеличению сывороточной концентрации карбамазепина, причем со степенью взаимодействия, связанного с дозой эритромицина [8/ Von Rosenstiel NA, Adam D. Macrolide antibacterials. Drug interactions of clinical signiÆcance. Drug Safety 1995; 13: 105±122]. У пациентов, получающих карбамазепин серьезные проявления токсичности происходят в пределах 3 дней от начала терапии эритромицином или кларитромицином. Механизм, лежащий в основе этого явления в ингибировании макролидами CYP 3A4 изоформы, для которой карбамазепин является [81/ Barzaghi N, Gatti G, Crema F, et al. Inhibition by erythromycin of the conversion of carbamazepine to its active 10,11-epoxide metabolite. Br J Clin Pharmacol 1987; 24: 936±938.] субстратом. Однако азитромицин и диритромицин свободны от взаимодействия с карбамазепином [42/ Watkins VS, Polk RE, Stotka JL. Drug interactions of macrolides: emphasis on dirithro-mycin. Ann Pharmacother 1997; 31: 349±356., 43/ Garey KW, Amsden GW. Intravenous azithromycin. Ann Pharmacother 1999; 33: 218±228.].
Антигистаминные средства.
Терфенадин - антигистамин, не обладающий седативным эффектом подвергается почти полной первичной биотрансформации и формированию окисленного метаболита посредством CYP 3A4. У восприимчивых индивидуумах, акумуляция основного состава может вызывать увиличение интервала QT, которое может закончиться трепетанием или мерцанием миокарда [82/ Kivisto KT, Neuvonen PJ, Klotz U. Inhibition of terfenadine metabolism: pharmaco-kinetic and pharmacodynamic
consequences. Clin Pharmacokinet 1994; 27: 1±5.]. Лоратадин, другой антагонист H1-рецепторов, метаболизируется в организме человека CYP 3A4 и в меньшей степени, CYP 2D6 [83/ Yumibe N, Huie K, Chen KJ, Clement RP, Caten MN. IdentiÆcation of human liver cytochrome P450s involved in the microsomal metabolism of the antihistaminic drug loratadine (Abstract). J Allergy Clin Immunol 1994; 93 (:1 Part;
2): 234.]. Однако, никакие изменения интервала QT и других побочных явлений при приеме лоратадина не наблюдалось. Carr и другие. [85/ Yumibe N, Huie K, Chen KJ, Clement RP, Caten MN. IdentiÆcation of human liver cytochrome P450s involved in
the microsomal metabolism of the antihistaminic drug loratadine (Abstract). J Allergy Clin Immunol 1994; 93 (:1 Part; 2): 234.] оценивали потенциал для взаимодействия между кларитромицином, назначаемым в дозе (500 мг) два раза в день, ежедневно, в течение 10 дней, и лоратадином в исследовании на здоровых добровольцах. Кларитромицин увеличил стационарный максимум наблюдаемой плазменной концентрации и AUC по интервалу дозирования для лоратадина (+36 % и +76 %) и для дезкарбоэтокси-лоратадина, активного метаболита лоратадина (+69 % и +49 %). Никакая связь между электрокардиографическими и фармакокинетическими взаимодействиями не наблюдалось. Астемизол, так же как и лоратадин подвергается первичному метаболизму до активных метаболитов и, подобно терфенадину, первичный состав обладает кардиотоксическим действием [82/ Kivisto KT, Neuvonen PJ, Klotz U. Inhibition of terfenadine metabolism: pharmaco-kinetic and harmacodynamic consequences. Clin Pharmacokinet 1994; 27: 1±5.].
Препараты спорыньи.
Наблюдаемый клинически эрготизм следовал из совместного применения препаратов спорыньи и эритромицина [87/ Ghali R, DeLean J, Douville Y, et al. Erythromycin-associated ergotamine intoxication: arteriographic and electrophysiologic
analysis of a rare cause of severe ischemia of the lower extremities and associated ischemic neuropathy. Ann Vasc Surg 1993; 7: 291±297.]. Этот неблагоприятный эффект приписывают ингибированию макролидами CYP 3A4, который метаболизирует препараты спорыньи. Клинический случай эрготизма с ишемией языка, вызванный взаимодействием кларитромицина и эрготамина был описан авторами [88/ Horowitz RS, Dart RC, Gomez HF. Clinical ergotism with lingual ischemia induced by clarithromycin±ergotamine interaction. Arch Intern Med 1996; 156: 456±458.
Macrolide-induced metabolic drug interactions f2000 Blackwell Science Ltd Br J Clin Pharmacol, 50, 285±295 295]. До сих пор, однако, никакой случай такого взаимодействия не был сообщен относительно азитромицина или диритромицина.
[Macrolide ± induced clinically relevant drug interactions with cytochrome P-450A (CYP) 3A4: an update focused on clarithromycin, azithromycin and dirithromycin .Jean Frederic Westphal 2000]
Следует отметить, что изоформа 3А принимает участие в метаболизме многих лекарственных препаратов применяемых в онкологии, в том числе и токсанов.
Токсаны.
Доцетаксел метаболизируется цитохромом CYP3A4. Таким образом, вн/видовая изменчивость активности CYP3A4 частично ответственна за различия в токсичности и клиренса. В єксперименте, проведенном авторами, двадцати одному пациенту в тяжелом состоянии, с диагнозом саркомы с наличием метастазов, без предыдущего лечения был назначен доцетаксел в дозе 100 мг/м2. Печеночная активность CYP3A4 у каждого пациента была измерена посредством [14C-N-метил] эритромицинового дыхательного теста (ERMBT). Пробы крови отбирались выборочно за следующие сутки после фармакокинетического анализа. Фенотипическая экспрессия печеночной CYP3A4 активности, измеренная ERMBT, была различна по 20 составляющим (применяли 14 C, выдыхая в течении 1 часа, среднее значение 2.53 %; диапазон, 0.25-5.35 %), что является подобным нормам контроля популяции . Клиренс доцетаксела различался, по 6 уровням показателей (средний, 21.0 л/ч/м2; диапазон, 5.4-29.1 л/ч/м2). ERMBT выявлялся лучшим предиктором клиренса доцетаксела, по сравнению с сывороточной аланин аминотрансферазой, альбумином, щелочной фосфотазой, или сывороточной альфа-1-кислым гликопротеином. Естественный уровень ERMBT составлял 67 % внутривидовых вариаций клиренса. Мультивариантный анализ показал, что естественный уровень ERMBT и альбумина вместе составляет 72 % внутривидовых вариаций в клиренсе. Самая высокая токсичность была отмечена у пациентов с самым низким ERMBT. Таким образом, активность CYP3A4 в печени самый мощный предиктор клиренса доцетаксела и составляет большинство его внутривидовых вариаций. [Malingre MM, Richel DJ, Beijnen JH, Rosing H, Koopman FJ, Ten Bokkel Huinink WW, Schot ME, Schellens JH Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel.J Clin Oncol 2001 Feb 15;19(4):1160-6 Related Articles, Books, LinkOut] Пациенты с низкой активностью CYP3A4 составляют группу риска (из за сниженного клиренса) и могут, таким образом, испытывать большую токсичность при приеме доцетаксела. Пациенты с высокой активностью CYP3A4 могут принимать субоптимальные дозы. Измеряя активность CYP3A4, ERMBT может быть клинически полезен в расчете доз субстратов CYP3A4, подобных доцетакселу. Совместное применение перорально циклоспорина, субстрата 3A4, значительно увеличивало биоэффективность доцетаксела. Средняя (+ /- SD) область под концентрационно-разовой кривой (AUC) у пациентов, которые получали перорально доцетаксел в дозе 75 мг/м2 без циклоспорина, была 0.37 + /- 0.33 мг.ч/л и 2.71 + /- 1.81 мг.ч/л для той же самой пероральной дозы доцетаксела с циклоспорином. Абсолютная биоэффективность перорального приема доцетаксела была 8 % + /- 6 % без, и 90 % + /- 44 % в комбинации с циклоспорином.[Hirth J, Watkins PB, Strawderman M, Schott A, Bruno R, Baker LH The effect of an individual's cytochrome CYP3A4 activity on docetaxel clearance Clin Cancer Res. 2000 Apr;6(4):1203-4.]
Как известно, кортизол метаболизируется цитохромом P450 (CYP3A4) и экскретируется с мочей, в виде 6-бета-гидроксикортизола (6beta-OHF) и свободного кортизола (FC), доцетаксел - также метаболизируется печеночным CYP3A4. Кортизол метаболизируется цитохромом P450 (CYP3A4) и экскретируется с мочей, в виде 6-бета-гидроксикортизола (6beta-OHF) и свободного кортизола (FC), доцетаксел - также метаболизируется печеночным CYP3A4. Данным методом авторами была изучена корреляция между фармакокинетикой доцетаксела и индивидуальной вариабельностью активности CYP3A4. После приема кортизола, общая сумма экскреции за 24 часа в моче 6бета-OHF (T6beta-OHF) резко увеличелась относительно исходного уровня (примерно в 60 раз), и составляла в среднем 12,273 + /- 4,076 мкг/d (означает + /- SD). Клиренс доцетаксела и область под концентрация-разовой кривой составляли в среднем 24.5 + /- 6.4 л/ч/м2 и 2.66 + /- 0.91 мг/л 8729. H. Превосходная корреляция наблюдалась между клиренсом доцетаксела и T6beta-OHF (r=.867). В многопрофильном анализе, T6beta-OHF (P < 001), альфа-1-кислый гликопротеин (P <004), AST (P = 007), и возраст (P = 022) знаменательно коррелировались с клиренсом доцетаксела.[Yamamoto N, Tamura T, Kamiya Y, Sekine I, Kunitoh H, Saijo N. LinkOutCorrelation between docetaxel clearance and estimated cytochrome P450 activity by urinary metabolite of exogenous cortisol. JClinOncol2000 Jun;18(11):2301-8 Related Articles, Books]
Органофосфатные пестициды.
CYP3A4 также играет важную роль в метаболизме органофосфатных пестицидов (OPs), например хлорпирифоса (Tanget al., 2001.) и паратиона О,О-диэтил-О-паранитрофенилтиофосфата (Butler and Murray, 1997; Eaton, 2000![]() ). Хлорпирифос - часто используемый OP инсектицид широкого спектра, который проявлят токсичность посредством ингибирования ацетилхолинэстеразы (Chambers,1992
). Хлорпирифос - часто используемый OP инсектицид широкого спектра, который проявлят токсичность посредством ингибирования ацетилхолинэстеразы (Chambers,1992 ![]() ). OPs ингибирует ацетилхолтнэстеразу, вызывая накопление нейромедиатора ацетилхолина в нервных и нервно-мышечных синапсах. Эти OPS используются как фосфоротиоат (P= S), который является очень слабым ингибитором ацетилхолинэстеразы. Однако, OPs преобразуется in vivo CYP ферментами из (P = S) до активного фосфатного эфира или оксона (P = O), который является мощным ингибитором ацетилхолтнэстеразы (Chambers, 1992).
). OPs ингибирует ацетилхолтнэстеразу, вызывая накопление нейромедиатора ацетилхолина в нервных и нервно-мышечных синапсах. Эти OPS используются как фосфоротиоат (P= S), который является очень слабым ингибитором ацетилхолинэстеразы. Однако, OPs преобразуется in vivo CYP ферментами из (P = S) до активного фосфатного эфира или оксона (P = O), который является мощным ингибитором ацетилхолтнэстеразы (Chambers, 1992).
Афлатоксин.
Shimada и Guengerich (1989) представили интересные доказательства о том, что главный катализатор, вовлеченный в биоактивацию гепатоканцерогена афлатоксина B (1) до его генотоксичной 2,3-эпоксид производной является нифидипин оксидаза, P450 белок, который также катализирует окисление нифидипина и других дигидропиридинов, квинидина, антибиотиков макролидов, различных стероидов, и других веществ. Уровни этого P450 фермента имеют большую амплитуду внутривидовых колебаний у людей, очевидно в широком унимодальном распределении (Schellens и другие., 1988). In vitro и in vivo было доказано, что проявление фермента может быть индуцировано барбитуратами, некоторыми стероидами, и макролидами. Так как активность фермента может быть оценена атравматичными исследованиями, стало возможным проверить гипотезу о связи рака печени с уровнем окислительного метаболизма в популяциях, в которых высоко потребление афлатоксина. Рак печени, главная причина преждевременной смерти в многих областях Африки и Азии, имеет инцидентность, которая строго коррелировала в областях с экспозицией к афлатоксину B(1). AFB (1) - микотоксин, производной разновидностью Aspergillus, и результатов экспозиции у человека преимущественно от потребления пищевых продуктов, загрязненных плесенью во время хранения. Канцерогенность их связана с конверсией до 8,9-оксида печеночным цитохром P450-зависимой системой монооксигеназ. Forrester и др. (1990) обнаружили, что уровни метаболической активации AFB (1) высоко коррелировали, и с уровнем белков CYP3A семейства генов и с полным содержанием в микросомах печени человека цитохрома P450.
Рассмотрим различные по механизму группы ингибиторов.
Макролиды, и подобные им ингибиторы относятся к группе быстро обратимых ингибиторов. Прямое быстро обратимое ингибирование есть закрепление ингибитора или его метаболита к CYP3A, оно было идентифицировано и являлось результатом конкурентного или неконкурентного ингибирования, степень которого определялась константой степени родства связи субстрата и ингибитора к ферменту и концентрации ингибитора. Критический фактор представлен соотношением ингибированного внутреннего клиренса в отсутствии ингибитора, так называемый "индекс ингибирования",- что есть отношение концентрации ингибитора к ее значению Ki [78, 79]. Таким образом, наиболее мощные обратимые CYP3A ингибиторы, которые включают азоловые противогрибковые препараты и первое поколение HIV ингибиторов протеаз, имеют значения Ki ниже 1 µM (Таблица 4). Другие лекпрепарпаты, известные как ингибиторы in vivo демонстрируют значения Ki, по меньшей мере в микромолярном диапазоне, явления мощного ингибирования не характерны для химических соединений со значениями Ki большее чем 75-100 µM (Таблица 4) потому что достаточно высокие уровни клинически не достигнуты.
Таблица 5 - Обратимые ингибиторы CYP3A- зависимого метаболизма человека.
| Ингибитор | Значения Ki (µM) | Механизм |
| Клотримазол | 0.00025–0.15 | |
| Кетоконазол | 0.015–8 | неконкурентноспособный, смешанный |
| итраконазол | 0.27 | |
| Миконазол | 0.9–1.3 | конкурентноспособный |
| Флуконазол | 1.3–63 | конкурентноспособный, неконкурентноспособный |
| Ритонавир | 0.017 | смешанный |
| Индинавир | 0.2 | конкурентноспособный |
| Саквинавир | 0.7 | конкурентноспособный |
| нелфинавир | 4.8 | конкурентноспособный |
| эритромицин | 16–194 | конкурентноспособный |
| тролеандомицин | 10–51 | конкурентноспособный, смешанный |
| джосамицин | 12–21 | Конкурентноспособный, смешанный |
| рокитамицин | 41 | |
| сертралин | 24–64 | смешанный |
| дезметилсертралин | 20–48 | смешанный |
| флуоксетин | 66–83 | смешанный |
| норфлуоксетин | 11–19 | смешанный |
| никардипин | 8 | конкурентноспособный |
| нифедипин | 10–22 | конкурентноспособный |
| верапамил | 24–82 | конкурентноспособный, смешанный |
| дилтиазем | 50–75 | конкурентноспособный |
| квинидин | 4–10 | конкурентноспособный |
| прогестерон | 8–45 | конкурентноспособный |
| дексаметазон | 23 | конкурентноспособный |
| этинилэстрадиол | 34 | смешанный |
| мидазолам | 40–63 | конкурентноспособный |
| циклоспорин | 1–37 | конкурентноспособный |
| рапамицин | 83 | конкурентноспособный |
| омепразол | 79 | конкурентноспособный |
| винбластин | 3.8 | смешанный |
| бромокриптин | 7–8 | конкурентноспособный |
| навелбин | 11 | смешанный |
| эрготамин | 12–14 | смешанный |
| кверцетин | 14 | неконкурентноспособный |
| винкристин | 19 | смешанный |
| дигидроэрготамин | 23 | конкурентноспособный |
Другая группа ингибиторов, к которой относятся некоторые 17-этинил замещаемые стероиды, например этинилэстрадиол, гестоден, и левоноргестрол, считаются необратимыми ингибиторами и инактивируют человеческий CYP3A в NADPH- и временно зависимом режиме [115, 116]. Антигестациозное средство, мифепристон (RU-486), также входило бы в эту категорию, но подтверждения данных о том, производит ли суицидальную инактивацию CYP3A у людей, еще не было получено. Недавно, экспериментальный препарат фуранопиридин, HIV ингибитор протеаз был признан высоко мощным и селективным по механизму ингибитором CYP3A [117], и это свойство было фактором, который сыграл отрицательную роль для фуранопиридина как кандидата на рынок лекарственных средств. Суммируя вышесказанное, возникает вопрос, обладают ли другие фураны подобными ингибирующими CYP3A свойствами. В этом отношении, интересно то, что 6 ', 7 '-дигироксибергамоттин, фуранокумарин, присутствуют в соке грейпфрута, прием которого может ингибировать первичный метаболизм CYP3A субстратов [118, 119]. Прием одного стакана сока грейпфрута демонстрировал значительное повышение оральной биодоступности различных медикаментов, часто используемых при лечении различных потологий, включая фелодипин, нифедипин, верапамил, этинилэстрадиол и циклоспорин A. Механизм этого эффекта состоит в мощном ингибировании метаболизма, что происходит ранее чем, абсорбция, так как многие из медикаментов, на которые воздействует сок, хорошо абсорбируюися и без присудствия сока грейпфрута. Большинство лекпрепаратов, на метаболизм которых воздействует сок грейпфрута, как известно, прежде всего метаболизируются CYP3A4, который является наиболее широко представленным цитохромом P450 в печени и энтероцитах, которые покрывают полость тонкой кишки. Lown и др. (1997) отметил, что главное местонахождение CYP3A4, на которое воздействует сок грейпфрута, представлено скорее тонкой кишкой, чем печенью. Т.е некоторые лекпрепараты, на которые воздействует сок грейпфрута сначала метаболизировались CYP3A4 в тонкой кишке. Кроме того, сок грейпфрута кажется, не влияет на клиренс CYP3A4 субстратов, когда они вводятся внутривенно. Наконец, первичный эффект сока грейпфрута при пероральном приеме медикаментов увеличивал пиковую серологическую концентрацию с небольшим изменением в последующей скорости элиминации, как измерено Т1/2. Lown и др. (1997) оценил эффект повторного приема сока грейпфрута на экспрессии CYP3A4 у 10 здоровых людей, которым давали 8 унций сока грейпфрута 3 раза в день в течение 6 дней. Они нашли, что сок грейпфрута не изменял уровней печеночной активности CYP3A4, а также ободочной кишки CYP3A5, или концентраций в тонкой кишке P-гликопротеида, а также, CYP1A1, и CYP2D6. Напротив, концентрация CYP3A4 в энтероцитах упала на 62 % без соответствующего изменения в CYP3A4 mRNA уровней. Кроме того, концентрации CYP3A4 энтероцитов, измеренные перед потреблением сока грейпфрута, коррелировали с увеличением пиковой серологической концентрации вовремя приема фелодипина с первым или шестнадцатым стаканом сока грейпфрута относительно воды. Lown и др. (1997) утверждают, что механизм эффекта сока грейпфрута на кинетику орального приема фелодипина в ее селективном снижении регуляции CYP3A4 в тонкой кишке. Не имелось никакого уменьшения эффекта сока грейпфрута через какое-либо время. Они наблюдали снижение регуляции CYP3A5 белка также как CYP3A4 белка. Кроме того, обнаружено, что этот эффект ассоциируется с аутокаталитической деструкцией кишечного контингента CYP3A, как in vitro [120], так и in vivo [121]. 6', 7'-дигидроксибергамоттин демонстрировал угнетение каталитической активности CYP3A in vitro [122].
Механизм суицидального ингибирования вовлекает CYP3A-зависимое формирование реактивного метаболита (ов), который в форме ковалентной связи с ферментом ведет к ее инактивации. Соответственно, в естественных условиях эффективность зависит от общей суммы, в большинстве от концентрации ингибитора, который подвергается экспозиции CYP3A; количества CYP3A; и порционирования реактивного метаболита между CYP3A и другими макромолекулами, относительно уровня ресинтеза нового фермента [114]. Поэтому, возможно, что, из-за меньшего количества CYP3A представленного в энтероцитах, в сравнении с печенью [64], механизм-основанное ингибирование после перорального приема, например, ацетиленовых стероидов селективно ингибирует скорее кишечный, чем печеночный метаболизм.
Интересными можно считать данные о взаимодействии CYP3A и Р-гликопротеида.
P-гликопротеид у людей – продукт MDR1 гена, который функционирует как трансмембранный насос. Его суперэкспрессия и роль в развитии мультилекарственной резистентности опухолевых клеток, подвергнутых экспозиции средствами химиотерапии, хорошо известны [17]. Однако, P-гликопротеид - также представлен в нескольких нормальных тканях [17], где рассматривается как функционирующий в системах: абсорбции кишечного эпителия, предотвращения распространения лекарственного средства (гематоэнцефалический барьер), или стимуляции элиминации лекарственного средства (печеночная каналикулярная мембрана и проксимальный почечный каналец). Таким образом, P-гликопротеид - колокализован в клетках, в которых CYP3A также экстенсивно экспрессируется, например энтероциты и гепатоциты. Эти два белка, функционируют в направлении контроля внутриклеточной концентрации ксенобиотиков. Известно, что значительное перекрытие спектров существует между химическими веществами и лекарственными препаратами, взаимодействующими с P-гликопротеидом и CYP3A [18]. Это вероятность случайна и скорее следует из широких специфик субстратов индивидуальных белков, чем из более фундаментальной взаимосвязи. Например, значительное число, но не все (например бензодиазепины), CYP3A субстраты взаимодействует с P-гликопротеидом как субстраты и-или ингибиторы (блокаторы кальциевых каналов, азол, противогрибковые средства, иммунодепрессанты и антибиотики с макроциклическим лактонным кольцом). Соответственно, коадминистрация двух CYP3A субстратов может иметь в результате взаимодействия, которые отражают ингибирование 1) только метаболизма, 2) только снижение P-гликопротеина, или комбинация обоих эффектов [19]. [Thummel K. E. In vitro and in vivo drug interactions involving HUMAN CYP3A. ,Annu. Rev. Pharmacol. Toxicol. 1998. 38:389-430]
Watkins в 1985 году идентифицировал глюкокортикоид-индуцибельный цитохром P450 в человеческой печени. Molowa в 1986 году сообщил о полной cDNA последовательности этого P450. Wrighton и Vandenbranden в 1989 изолировали CYP3-тайп цитохрома P450 из человеческой плодной печени. Гибридизацией соматической клетки и гибридизацией in situ, Riddell и в 1987 году определил, что ген, кодирующий фермент нифидипин- оксидазу (CYP3) находится на 7 хромосоме. Назначение 7 хромосомы было подтверждено Gonzalez в 1988 году методом использования гибридов соматической клетки. Эти авторы также обеспечили дополнительное доказательство в поддержку тождества P450PCN1 и нифидипин- оксидазы. Многоточечным анализом связи, использующим DNA маркеры, с известным расположением на хромосоме 7, Brooks в 1988 году пришел к заключению, что наиболее вероятное месторасположение CYP3 является- 7q21-q22.1. Никакая рекомбинация с COL1A2 (120160) маркером не была найдена. Spurr в 1989 году определил CYP3 на 7q22-qter изучением набора гибридов соматической клетки по принципу человек- грызун. Inoue в 1992 году отобразил CYP3A4 к 7q22.1 флюоресценцией гибридизации in situ. Далее, Lehmann в 1998 году идентифицировал отдельный рецептор ядра человека, названный прегнан X рецептором (PXR), который связывается с элементом ответа в CYP3A4 промоторе и активизируется широким диапазоном лекарственных препаратов, известных как идукторы экспрессии CYP3A4. При сравнении человеческого PXR с Pxr мыши получены различия в их активации некоторыми медикаментами, которые можно частично объяснить видоспецифичными эффектами соединений на экспрессию CYP3A4 гена. Эти результаты обеспечили возможность объяснения на молекулярном уровне способности неполярных химических соединений индуцировать CYP3A4 и, кроме того, обеспечили основание для разработки в исследованиях in vitro, возможностей предикции взаимодействия медикаментов у человека. Индукция CYP3A ферментов видоспецифична и включает в себя 1 и более клеточных факторов или рецептор-подобных ксеносенсоров.
Рис.10 - Механизм индукции генов цитохрома Р-450 с учетом роли ядерных рецепторов и других внутриклеточных систем
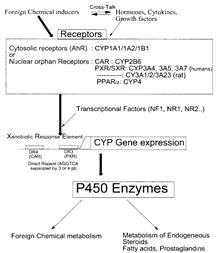
Xie в 2000 году идентифицировал PXR/SXR как один из таких факторов. Он показал что целенаправленный сбой Pxr гена мышей блокировал индукцию CYP3A прототипами индуктора типа дексаметазона или прегненолон-16-альфа-карбонитрила. У Pxr-свободных мышей, несущих трансген для активизированной формы человеческого SXR, с конститутивно высоким уровнем экспрессии CYP3A гена, происходило усиление протекторной функции относительно токсических ксенобиотиков. Xie также продемонстрировал, что разновидности начал рецептора, скорее всего представляют собой структуру промотора CYP3A генов, который отвечает за видоспецифичный образец индукции CYP3A. Таким образом, появилась возможность генерировать 'гуманизированных' мышей, содержащих трансген, которые были чувствительны к человеческим специфическим индукторам типа антибиотика рифампицина. Xie сделал заключение, что SXR/PXR гены кодируют первичные видоспецифичные ксеносенсоры, которые являются посредниками адаптивного печеночного ответа, и могут представлять основу биохимического механизма человеческой ксенопротекции. [N. Engl. J.Med 338, 916, 1998; Drug Metab. Dispo 27, 1260, 1999]. Tirona в 2003 году доказал, что отдельный нуклеарный рецептор гепатоцита нуклеарный фактор -4-альфа (HNF4A; 600281) вовлечен в PXR- и CAR-опосредованную активацию транскрипции CYP3A4. Он идентифицировал специфический цис-действующий элемент энхансера CYP3A4 гена , который способствует закреплению HNF4-альфа и таким образом способствует PXR- и CAR-опосредованной активации гена. Фетальные мыши с условным делетированным Hnf4-альфа демонстрировали снижение или отсутствие экспрессии CYP3A. Кроме того, взрослые мыши с условно делетированным геном в тканях печени демонстрировали снижение базальной и индуцированной экспрессии CYP3A. Эти данные идентифицировали HNF4-альфа как важный регулятор координации реакции на ксенобиотики, опосредованной нуклеарным рецептором.
Рис 11 - Роль ядерных рецепторов в процессе транскрипции гена
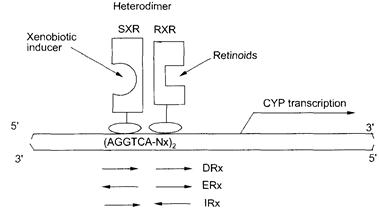
Как извесно, два близко родственных самостоятельных гормональных рецептора ядра – вышуказанный прегнан X рецептор (PXR) и основной андростан рецептор (CAR) - появились как регуляторы транскрипции и экспрессии гем- содержащих монооксигеназ цитохрома P 450, с которыми связываются, подверженные окислительному метаболизму ксенобиотики. [Clin. Pharmcokinet 38, 493, 2000].
Таблица 6 - Механизмы индукции различных изоформ цитохрома Р-450
| Механизм индукции | Индукция и регуляция P450 |
| Транскрипця генов посредством рецепторов | 1A1 (цитозоль AhR), 1A2, 1B1 2A6, 2B6 (CAR), 2C8, 2C9, 2C18, 2C19 3A4, 3A5 (ядерные рецепторы PXR и SXR),4A11 (PPARa) |
| mRNA процессинг | 1A2 |
| mRNA стабилизация | 1A1,2E1,3A4 |
| Ферментная стабилизация | 2E1 |
Открытие PXR как первичного регулятора индукции экспрессии CYP 3A в печени и кишнике имеет большое значение для процессов биотрансформации лекарственных средств [J. Clin. Invest, 102, 1016, 1998] Как известно, посредством CYP3A метаболизируется большинство лекарственных препаратов, и ненамеренная активация PXR у людей может приводить к нежелательным взаимодействиям лекарственных средств или увиличения уровней токсических метаболитов.[Clin. Pharmcokinet 38, 493, 2000].[Bertilsson et al. 1998, Blumberg et al. 1998, Lehmann et al. 1998]. Примечательно, что экспрессии PXR не было обнаружено авторами в тканях человеческого легкого методом Northern blot analysis [Bertilsson и другие. 1998, Blumberg и другие. 1998, Lehmann и другие. 1998]. Что, однако не исключает возможности экспрессии в некоторых специфических типах легочных клеток. [Baes et al. 1994]. Чрезвычайно низкие уровни САR mRNA обнаружены в легочной ткани методом Northern blot после длительной экспозиции [Baes и другие. 1994].[Lampen A, Christians U, Guengerich FP, Watkins PB, Kolars JC, Bader A, Gonschior AK, Dralle H, Hackbarth I, Sewing KF: Metabolism of the immunosuppressant tacrolimus in the small intestine: Cytochrome P450, drug interactions, and interindividual variability.], [Drug. Metab. Dispos 1995, 12:1315-1324, Olsen A, Hansen KT, Friis C: Pig hepatocytes as an in vitro model to study the regulation of human CYP3A4: prediction of drug-drug interactions with 17І-ethynylestradiol.], [Chem.-Biol. Interact 1997, 107:93-108, Bader A, Hansen T, Kirchner G, Allmeling C, Haverich A, Borlak JT: Primary porcine enterocyte and hepatocyte cultures to study drug oxidation reactions.Br. J. Pharmacol 2000, 129:331-342].
Поскольку человеческий CYP3A P450 семейство, состоящее по крайней мере из четырех высоко гомологичных генов, и экспрессируется восновном в печени. Для того, чтобы исследовать экспрессию членов семейства CYP3A индивидуально, авторы подготовили олигонуклеотиды, специфичные к каждому CYP3A mRNA и использовали Северный блот-анализ и/или полимеразную цепную реакцию, для изучения РНК взрослой и плодной печени, вариации в экспрессии форм CYP3A в течение развития. Schuetz JD и др., нашли, что значения CYP3A4 (P450NF) mRNA, были получены только Северным блотом в постнатальном периоде, отличались высокой вариабельностью (с 10 уровнями) посравнению с образцами взрослых, и, в отличие от копий крыс (CYP3A1/2), не находились под влиянием рода. Напротив, присутствие изоформы CYP3A7 (HFLa) mRNA, как предполагалось, не было ограничено плодной печенью, а наблюдалось в семи из 13 образцов печени взрослых (54 %). CYP3A5 (HLp2), как mRNA, также был обнаружен во всех образцах плодной, и в трех из 13 взрослой печени (23 %) а в двух из этих трех соэкспрессировался с CYP3A7 mRNA.
CYP3A5 и CYP3A7 экспрессируются на сходных уровнях в плодной печени любого рода. Кроме того, экспрессия CYP3A7 в плодной печени имеет меньшее значение переменной (< с 2.5 уровней) чем CYP3A4 у взрослых. Авторы утверждают, что вопреки преобладанию представлений, экспрессия CYP3A7 в печени не ограничевается фетальным периодом, а скорее представляет вторую CYP3A форму, которая селективно экспрессируется у взрослых. [Selective expression of cytochrome P450 CYP3A mRNAs in embryonic and adult human liver. Schuetz JD, Beach DL, Guzelian PS. Department of Medicine, Medical College of Virginia, Richmond 23298-0267.]
Используя искусственный бактериальный хромосомный (BAC) клон, Finta C. и др. отобразили локус человеческого цитохрома P450 3A (CYP3A), содержащий гены кодирующие CYP3A4, CYP3A5 и CYP3A7. Гены лежат по направлению "голова® хвост" в порядке 3A4, 3A7 и 3A5. В обоих межгенных областях (3A4-3A7 и 3A7-3A5), авторы обнаружили несколько дополнительных экзонов цитохрома P450 3A, формирующих два CYP3A псевдогена. Эти псевдогены имеют ту же самую ориентацию как CYP3A гены.. Удивляет, что 3A7 mRNA разновидность была обнаружена между экзонами 2 и 13 одного из псевдогенов (нижележащего по отношению к 3A7) которые соединены после 3A7 терминала экзона. Это происходит как в mRNA молекуле, которая состоит из 13 3A7 экзонов и двух дополнительных экзонов в 3 ' конце. Дополнительные два экзона, берущих начало от псевдогена, находятся в измененной рамке считывания и следовательно имеют способность кодировать полностью отличную последовательность аминокислоты, чем канонические CYP3A экзоны 2 и 13. Эти результаты могут представлять генерализованный эволюционный процесс, происходящий с генами, имеющими потенциал, для захвата (кэпирования) соседних последовательностей и использования их как функциональных экзонов. [Finta C, Zaphiropoulos PG ,The human cytochrome P450 3A locus. Gene evolution by capture of downstream exons.. Center for Nutrition and Toxicology, Department of Biosciences at NOVUM, Karolinska Institute, SE-14157, Huddinge, Sweden.] Имеется приблизительно 10 уровневая вариабельность метаболизма CYP3A4 субстратов in vivo, включая антибиотики рифампицин и кетокеоназол, блокатор кальциевых каналов нифидипин, и иммунодепрессант циклоспорин [Thummel и Wilkinson, 1998; Guengerich, 1999]. Эта вариация может затрагивать эффективность лекарственного средства и токсичность. Вариабельность экспрессии CYP3A4 по крайней мере частично зависит от множественных факторов, включая индукцию лекпрепаратами, эндогенными соединениями, и химикалиями окружающей среды, а также включает генетические факторы. Последние данные свидетельствуют о том, что область кодирующая CYP3A4 - также наследственно вариабельна [Sata и другие., 2000; Eiselt и другие., 2001].
Особенности генетических вариаций CYP3A4 недавно стали извесны. Мутация в 5'-upstream боксе проявлялась CYP3A4*1B (A290G), и наблюдалась в 52 % афроамериканцев и 9.6 % европейцев, но не была идентифицирована у азиатов [Ball et al., 1999; Rebbeck,2000; Sata et al., 2000; Gellner et al., 2001]. Это предположение было связано с прогрессирующей стадией рака простаты у людей [Rebbeck и другие., 1998], и тем не менее несет защитные эффекты для вторичного рака, вызванного препаратами химиотерапии, типа эпиподофилотоксинов (которые метаболизируются CYP3A4) используемых для лечения лейкемии,. [Felix et al., 1998.]. Однако, этот полиморфизм кажется, не затрагивает конститутивных уровней CYP3A4 [Wandel и другие., 2000]. Gonzales и коллеги [Sata и другие., 2000] описали два кодирования SNPs, включая CYP3A4*2 (S222P) найденный только у европейцев финского происхождения с аллельной частотой 2.7 %, и единственный случай CYP3A4*3 (M445T) в китайской популяции из 178 индивидуумов.
Baculovirus, экспрессирующий CYP3A4*2 белок, демонстрировал увеличение в Km для нифидипина, но не для тестостерона, в отличии от CYP3A4*1. Что ограничивало информативность эффектов новой M445T аллели на метаболизм [Sata и другие., 2000]. CYP3A4*4 (I118V), CYP3A4*5 (P218R), и CYP3A4*6 (терминирующий кодон в 285) были получены авторами в китайской популяции с аллельными частотами 1.4%, 0.98%, и 0.5%, и все эти различные аллели были связаны с более низкими соотношениями 6 ![]() -гидроксикортизола к свободному кортизолу, как и в исследованиях in vivo. [Hsieh и другие., 2001]. Каталитическая активность неизмененного типа CYP3A4 и аллельных вариантов сравнивались авторами, используя тестостерон и инсектицид хлорпирифос, как прототипы 3A4 субстратов. Две аллели, S222P и L373F, как стало извесно , оказывают влияние на каталитическую активность к некоторым субстратам. [Sata и другие., 2000
-гидроксикортизола к свободному кортизолу, как и в исследованиях in vivo. [Hsieh и другие., 2001]. Каталитическая активность неизмененного типа CYP3A4 и аллельных вариантов сравнивались авторами, используя тестостерон и инсектицид хлорпирифос, как прототипы 3A4 субстратов. Две аллели, S222P и L373F, как стало извесно , оказывают влияние на каталитическую активность к некоторым субстратам. [Sata и другие., 2000 ![]() ; Eiselt и другие., 2001]. Интересно, F189S и L293P мутации имели сходное влияние на метаболизм обоих субстратов подобным образом. F189S аллель значительно понижает интенсивность обмена для тестостерона и хлорпирифоса, по сравнению с неизмененным типом CYP3A4, в то время как L293P аллель продемонстрировала более высокие значения обмена для обоих субстратов. Эти результаты свидетельствуют об возможности индивидуальных изменений способности метаболизировать не только тестостерон, но и другие потенциальные субстраты.
; Eiselt и другие., 2001]. Интересно, F189S и L293P мутации имели сходное влияние на метаболизм обоих субстратов подобным образом. F189S аллель значительно понижает интенсивность обмена для тестостерона и хлорпирифоса, по сравнению с неизмененным типом CYP3A4, в то время как L293P аллель продемонстрировала более высокие значения обмена для обоих субстратов. Эти результаты свидетельствуют об возможности индивидуальных изменений способности метаболизировать не только тестостерон, но и другие потенциальные субстраты.
Значения обмена для CYP3A4*1, F189S, L293P, M445T, и P457S 7.03, 1.9 (P < 0.05), 12.4 (P < 0.05), 5.8 и 5.9 nmol/min/nmol.
Сообщения о последних результатах исследований обеспечили обстоятельное свидетельство в поддержку ассоциации между генетическим полиморфизмом (A->G) в 5 '-фланкированным регионом (-290) CYP3A4 и изменениями ферментной активности. [. P. Rivory (1)(2), H. Qin (3), S. J. Clarke (2), J. Eris (4), G. Duggin (4), E. Ray (4), R. J. Trent (3), J. F. Bishop (2)Frequency of cytochrome P450 3A4 variant genotype in transplant population and lack of association with cyclosporin clearance., L]Как известно CYP3A активность есть сумма активностей семейства CYP3A генов, включая CYP3A5, для которого характерен полиморфизм экспрессии на высоком уровне. Поскольку CYP3A5 представляет по крайней мере 50 % от общего печеночного содержания CYP3A, полиморфизм его экспрессии у людей имеет огромное значение, поэтому CYP3A5 может быть наиболее значимым генетическим контрибьютером к межиндивидуальным и межрасовым различиям в случаях CYP3A-зависимого лекарственного клиренса и в качестве ответной реакции на введение многих лекарственных препаратов. [Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E, Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression.. Department of Molecular and Cell Biology, University of Maryland at Baltimore, Baltimore, Maryland, USA]
Как стало недавно известно, экспрессия CYP3A5 в альвеолярных макрофагах репрессируется курением табака [Piipari и др. 2000]. Механизм этого явления еще недостаточно изучен.
Люди имеющие, по крайней мере, одну аллель CYP3A5*1 экспрессируют относительно большие количества CYP3A5. Результаты, представленные авторами, указывают на то, что полиморфизм одного нуклеотида (SNPs) в CYP3A5*3 и CYP3A5*6 аллелях, в результате альтернативого сплайсинга и белковой трансации заканчивается отсутствием CYP3A5 в человеческих тканях у некоторых индивидуумов. CYP3A5 чаще экспрессируется в печени афроамериканцев (60 %) чем в у евроамериканцев (33 %). [Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E.,Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Department of Molecular and Cell Biology, University of Maryland at Baltimore, Baltimore, Maryland, USA].
Отличия в активности различных ферментов цитохрома P450 (CYP) в популяциях демонстрировались авторами на основе генетических или энвироментальных детерминат. Интересными можно считать изучения, направленные на выяснение различий и особенностей метаболической активности различных изоформ иих аллелей у разных демографических групп. Испанцы - большая демографическая группа, и во всем мире и в пределах Соединенных Штатов; следовательно возможность различий в метаболизме между мексико и евро- производными популяциями было бы интересно определить относительно CYP2E1 и CYP3A. Авторы провели исследование, где все 15 субъектов, принимавших участие в исследовании были мужчины и получали внутривенно в дозе 1 мг и орально в дозе 2 мг [(15) N (3)] -маркированный мидазолам единовременно для того, чтобы характеризовать диспозицию бензодиазепина в этих двух популяциях. Отсутствие любых различий в диспозиции мидазолама указывает но то, что, с фармакокинетической точки зрения, дозировки лекпрепаратов, метаболизируемых CYP3A не должны иметь различия у мексико и евроамериканцами. [Cytochrome P450 2E1 and 3A activities do not differ between Mexicans and European Americans.Poland RA, Lin KM, Nuccio C, WilkinsonGR. Department of Psychiatry, Research Center on the Psychobiology of Ethnicity, Harbor-UCLA Medical Center, Torrance, CA, USA.]
Экспрессия CYP3A4 изменяется по 40 уровням в зависимости от индивидуальных собенностей человеческой печени, и метаболизм CYP3A4 субстратов изменяется по крайней мере по 10 уровням in vivo. Полиморфизмы одного нуклеотида (SNPs) в CYP3A4 были идентифицированы прямым секвенированием геномного DNA в 72 индивидуальных формах от трех различных этнических групп, включая европейцев, негроидов (афроамериканцы и африканские пигмеи), и представителей азии. Общее количество из 28 SNPs было идентифицировано, авторами, включая пять отвечающих за кодировку изменений M445T (CYP3A4*3), R162Q (CYP3A4*15), F189S (CYP3A4*17), L293P (CYP3A4*18), и P467S (CYP3A4*19). Последние четыре представляют новые варианты аллелей. Расовая вариабильность наблюдалась для частоты индивидуального SNPs. CYP3A R162Q был идентифицирован только в негритянских популяцияхях с аллельной частотой 4 %. CYP3A4 F189S и CYP3A4 M445T были идентифицированы у европейцев с аллельными частотами 2 % и 4 %, соответственно. L293P и P467S наблюдались только у азиатов с аллельной частотой 2 %. сDNAs для F189S, L293P, M445T, и P467S мутантных аллелей были получены сайт-направленным мутагенезом и экспрессированы на системах экспрессии Escherichia coli. Тестостерон и инсектицид хлорпирифос использовались авторами для оценки каталитической активности наиболее общей CYP3A4 аллели (CYP3A4*1) и ее аллельных вариантов. CYP3A4 F189S демонстрирует более низкие показатели обмена для тестостерона и хлорпирифоса, в то время как CYP3A4 L293P имел более высокие значения обмена для обоих субстратов. Показатели обмена CYP3A4 M445T и P467S аллелей, метаболизирующих вышеуказанные соединения не имели достоверных различий от таковых неизмененного типа CYP3A4. [Anzenbacher P, Soucek P, Anzenbacherovб E, Gut I, Hrubэ K, Svoboda Z, Kvetina J: Presence and activity of cytochrome P450 isoforms in minipig liver microsomes. Comparison with human liver samples.Drug Metab. Dispos 1998, 26:90-93].
Огромное клиническое значение имеют возможности выявления предикторов вторичных новообразований. Поскольку только у меньшинства пациентов с лейкемией возникшей после химиотерапии, различия во взаимодействиях лекарственных средств могут иметь множество предрасполагающих факторов. Мутации в тумор-супрессорных генах или генетических вариациях в метаболизме лекарственного средства - примеры основных факторов риска. Мутации в NF1 (162200) и p53 (191170) тумор-супрессорных генах наблюдались в случаях лекемии связанной с приемом алкилирующих цитостатиков и моносомиями хромосом 5 и 7. Избыток GSTT1 (600436) свободного генотипа наблюдался во взрослой белой популяции с миелодиспластическим синдромом, который снижал детоксикацию канцерогенных веществ, и соответственно, расширял восприимчивость к миелодиспластическому синдрому [Chen и другие., 1996]. Эпиподофилотоксины, которые используются как ингибиторы DNA топоизомеразы 2 в лечении лейкемии связаны с продукцией транслокаций, включая MLL ген, за счет субстратов метаболизма CYP3A. Paolini в 1999 году сообщил о существенных увеличениях канцероген- метаболизирующих ферментов CYP1A1 (108330), CYP1A2, CYP3A, CYP2B (123930), и CYP2A в легких крыс, дополненных высокими дозами бета-каротина. Авторы утверждают, что соответственно высокие уровни CYPS у людей предраспологают индивидуума к риску развития рака при биоактивации табачным дымом широкого спектра проканцерогенов, таким образом объясняя коканцерогеный эффект бета-каротина у курильщиков. Имеются несколько проводящих путей, вовлеченные в метаболизм тестостерона, и генов, которые регулируют эти проводящие пути, включая 5-альфа-редуктазу-2 (SRD5A2; 607306) и CYP3A4, которые были вовлечены в пороцессы восприимчивости при раке простаты (176807). CYP3A4*1B аллель (124010.0001) может уменьшать окислительную деактивацию тестостерона [Rebbeck и другие., 1998]. Известно, что афрамериканцы имеют самый высокий зарегистрированный уровень рака простаты в мире. Zeigler-Johnson в 2002 году изучали различия в генотипах SRD5A2 и CYP3A4 согласно этнической принадлежности. Они нашли, что CYP3A4*1B аллель чаще встречалась у жителей Ганы и афроамериканцев, (частота гена больше чем 50 %) чем у европейцев (меньше чем 10 %), и очевидно отсутствовала у азиатов. Rebbeck в 1998 году наблюдал полиморфизм в нифидипин-специфичном элементе ответа CYP3A4 промотора, которым он определил как CYP3A4-V. Уокер в 1998 году обнаружил, что последовательность agggcaagag была наиболее часто встречаемой у европейцев и тайванцев; agggcaggag (CYP3A4-V) присутствовал у 9 % европейцев, 53 % афроамериканцев, и 0 % тайванцев. Существенный дефицит изоформы CYP3A4-V был найден Феликсом в 1998 году у субъектов, которые страдали связанной с предыдущим химиотерапевтическим лечением лейкемией, при условии, что лечение проводилось препаратами, которые метаболизируются посредством CYP3A.
Человеческие раковые образования ободочной кишки, груди, легкого, печени, почек и простаты, как известно, также экспрессируют изоформы цитохрома P450 (CYP) , включая изоформы CYP 3A и CYP 1A .
Изучение гепатоцелюлярных опухолей методом иммуногистохимического окрашивания демострирует наличие CYP1 в большинстве (65 %) а также CYP 3A в почти половине 42 % исследований. Другие исследования показали, что первичные и вторичные опухоли печени могут экспрессировать CYP 450 изоформы 1A1, 2A6. 2B6, 2C8/9. 3A4, 4A. [Philip ft al., 1994]. Более детальное изучение показало, что и CYP IA и CYP3A белок присутствовал во всех аденомах и > 60 % раков. [McKay et til.. 1993]Рак желудка также экспрессирует CYP1A (51%i) и CYP3A (28%i). [Murray i't cil.. 1998]. Клетки рака почки, основной злокачественной опухоли почки, последовательно экспрессирует CYP3A. RT-PCR показал, что CYP3A5 и CYP3A7 mRNA представлены во всех образцах опухоли, тогда как CYP3A4 mRNA был обнаружен в большинстве (65 %,)[Murray ft ill.. 1999]. Аденокарциномы легких и клетки карциномы, все экспрессировали CYP3A, тогда как CYP2E1 был представлен только в (46 %.).
Суммируя все вышеизложенное, необходимо отметить, что в практическом аспекте важно учитывать то, что пути метаболических превращений лекарственных средств и пути метаболических превращений токсичных соединений носят однонаправленный характер, поэтому на путях метаболизма лекарственных средств и ксенобиотиков возможно развитие реакций взаимодействия. Имеется достаточное количество экологических факторов, угнетающих активность Р 450 - это ацетат ртути, свинец, никель, кадмий, мышьяк, фенолы, четыреххлористый углерод, анилин. Напротив, к стимуляторам цитохрома Р 450 относятся бензопирен, ацетон, бензол, изопропанол, хлорированные инсектициды (ДДТ, гексахлоран). Способностью повышать активность пероксидазы, НАДФ - цитохром Р450 - редуктазы обладают соединения марганца. На активность флавопротеинов избирательно оказывают стимулирующее действие марганец, железо, медь, молибден.
Поэтому явления профессиональных вредностей, а так же состояние экологической ситуации, ее влияние на генетический механизм, так же необходимо учитывать.
Похожие работы
... м, в течение 10-15 мин. Группа низкоустойчивых (НУ) животных выдерживала эту высоту только в течение 1-1.5 мин. Глава 5. Результаты и обсуждения 5.1 Параметры функционирования митоКАТФ канала у крыс с различной резистентностью, а также у животных, адаптированных к гипоксии В этом разделе работы исследовались такие показатели, как дыхание МХ, скорость АТФ-зависимого К+ транспорта, ...
0 комментариев