Реферат
Тема: ФАРМАКОКИНЕТИЧЕСКИЕ ФАКТОРЫ
В АНЕСТЕЗИОЛОГИИ
План
Вступление
Общая фармакокинетика
Фармакокинетические факторы
Фармакокинетика средств, применяемых в анестезиологии
Список литературы
Вступление
Всегда и в любых ситуациях анестезиолог действует прежде всего как клинический патофизиолог и клинический фармаколог. Клинической физиологии и патофизиологии посвящены многие главы руководств. В этой работе особое внимание уделено клинической фармакокинетике, которая занимает незаслуженно малое место в учебниках и руководствах по анестезиологии и реаниматологии.
Общая фармакокинетика
Грамотное применение медикаментозных средств, выбор их дозы, времени, пути и способа введения для получения нужного эффекта требует от анестезиолога-реаниматолога знания и понимания прежде всего основополагающих принципов фармакологии. Чисто прагматический подход приводит к тому, что на первый план выступает интерес к конечному результату - специфическому фармакологическому действию препарата, которое обусловлено его свойствами и созданием в организме (в крови, органах и тканях) определенной его концентрации. Эта зависимость между концентрацией лекарственного средства и вызываемым им эффектом служит предметом фармакодинамики. Но для создания эффективной концентрации необходимо, чтобы в организм поступила определенная доза препарата Зависимость между дозой и концентрацией лекарственного средства в крови и других жидкостях организма является объектом фармакокинетики.
Суть фармакокинетики заключается в приложении кинетики (т.е. скорости изменения в любой системе) к фармакону (греч pharmakon лекарство) Иными словами, фармакокинетика - это скорость и характер изменении концентрации медикаментозного средства в биологической системе - организме и его составляющих (органы, ткани, жидкости).
Таким образом, фармакология состоит из двух частей - фармакодинамики и фармакокинетики.
Если фармакодинамика (пусть даже в эмпирической форме) была руководством к действию, очевидно, на протяжении всей истории существования человечества, то фармакокинетика как раздел науки достаточно молода.
Зависимость между дозой лекарства и его терапевтическим или токсическим эффектом была известна уже со времен Парацельса (XVI в). В 1866 г. О. Шмидеберг свою диссертацию на соискание ученой степени доктора медицины посвятил аналитическому методу определения содержания хлороформа в крови. Но становление фармакокинетики как науки произошло только в первой половине нынешнего столетия. В значительной степени ее прогресс был связан с привлечением математического аппарата (возможно, это обусловило ее меньшую по сравнению с фармакодинамикой популярность у клиницистов). Термин "фармакокинетика" впервые предложил Ф. Дост в 1953 г. Но только в 70-80-х годах появились первые монографии, посвященные фармакокинетике [Соловьев В.Н. и др., 1980; Холодов Л.Е., Яковлев В.П., 1985; Wagner J. G., 1975; Gibaldi M., Perrier D., 1975; Stanski D. R., Watkins W. D., 1982; Prys-Roberts С., Hug С.С., 1983].
Фармакокинетические факторыФармакокинетические факторы определяют скорость изменения концентрации лекарственного соединения в период или после его применения и, следовательно, детерминируют наступление, интенсивность и продолжительность эффекта препарата. Эти факторы включают все процессы, которые могут повлиять на фармакологический препарат: абсорбцию в кровь в области введения, распределение по органам и тканям, элиминацию путем метаболизма или экскреции. Иначе говоря, фармакокинетика изучает судьбу лекарственного препарата, его путь от момента введения (или приема) до полного выделения или распада.
Кратко рассмотрим эти процессы. Поступление препарата возможно несколькими основными путями (прием внутрь, введение подкожно, внутримышечно и внутривенно, ингаляционным путем). Учитывая специфику использования лекарственных средств в анестезиологии, особое внимание уделим инъекционному и ингаляционному способам их введения.
Скорость абсорбции лекарственного средства при подкожном введении зависит от васкуляризации тканей: вазоконстрикция уменьшает абсорбцию, вазодилатация в месте инъекции - увеличивает. Эти условия действуют также при внутримышечной инъекции, причем абсорбция в этом случае более быстрая, чем при подкожном введении, особенно при использовании водорастворимых препаратов. Темп абсорбции падает в случае применения лекарственных препаратов в виде эмульсии или растворенных в масле. Эти формы фармакологических средств используют специально для обеспечения их медленного постепенного всасывания и длительного эффекта. Та часть препарата, которая попадает в кровоток, получила название доступной фракции. При внутривенной инъекции препарат полностью попадает непосредственно в кровоток.
Различия скорости абсорбции препарата в крови обусловливают неодинаковые изменения его концентрации при том или ином пути введения. После приема внутрь концентрация, медленно повышаясь, достигает пика, а затем постепенно снижается. Быстрее и значительнее высокий пик концентрации создается при подкожном введении. Следует обратить внимание на то, что высокая начальная концентрация препарата в крови после внутривенной инъекции быстро падает.
Соответственно изменениям концентрации в крови меняется и интенсивность фармакологического действия. После внутривенной инъекции эффект наступает более быстро, но его продолжительность меньше, чем при любом другом пути введения. Варьируя скорость введения, можно изменить и действие препарата, вводимого внутривенно. При быстрой внутривенной инъекции не успевает произойти разведение препарата кровью, что приводит к поступлению в ткани лекарственного средства в высокой концентрации и быстрому развитию фармакологического эффекта. Последующее столь же быстрое ослабление этого действия обусловлено главным образом падением концентрации в крови в связи со смешиванием, а не быстрым начальным перераспределением в депо. Постоянный уровень препарата в крови может быть обеспечен длительной внутривенной инфузией с определенной скоростью.
Ингаляционный путь введения фармакологического средства характеризуется быстрой абсорбцией через легкие, что обусловлено большой поверхностью всасывания и интенсивным кровотоком. Эффективность ингаляционного пути почти столь же велика, как и внутривенной инъекции.
Анализируя особенности абсорбции лекарственных препаратов при различных путях введения, мы уже упоминали ряд факторов, которые влияют на концентрацию медикамента и его распределение в организме. Распределение препарата между различными секторами организма начинается фактически с момента его поступления. На этот процесс влияют скорость кровотока и степень кровоснабжения органов и тканей, связывание препарата с белками крови, депонирование в жировых и иных депо, метаболизм и распад, выделение из организма почками, печенью, легкими, с калом. Обсуждение этого вопроса начнем с рассмотрения ряда процессов и понятий, необходимых для понимания кинетики фармакологических средств.
После абсорбции из места введения (а после внутривенной инъекции непосредственно) лекарственный препарат попадает в кровоток, где в течение нескольких кругооборотов крови происходит смешивание или растворение медикамента в плазме. Препараты с кислой и основной реакцией находятся в плазме в ионизированной и неионизированной формах, соотношение которых зависит от рН и определяется уравнением Гендерсона-Хассельбальха. Так, при нормальном рН крови (7,4) тиопентал-натрий ионизирован на 30%, а диазепам - только на 0,01%. Это обстоятельство имеет существенное практическое значение, так как только неионизированная форма фармакологического средства проникает через липидные мембраны или растворяется в жировых соединениях, поэтому ее можно считать активной диффундирующей фракцией. Поскольку существует очень чувствительное, быстро восстанавливаемое равновесие между ионизированной и неионизированной формами, ионизация не влияет на распределение препарата между тканями с равным рН, а сказывается только на его скорости.
Проникновение в клетки (например, эритроциты) зависит от растворимости в жирах. Так, фентанил, будучи высокорастворимым в жировых субстанциях, свободно проникает в эритроциты, а плохо растворимый в жирах панкуроний не проникает.
Многие препараты в крови связываются с белками, главным образом с альбуминами. Это связывание носит динамический обратимый характер. Каждая молекула белка может иметь несколько точек связывания, аффинитет которых к одному и тому же препарату может быть различным.
Связывание и диссоциация происходят очень быстро и практически постоянно. По мере увеличения концентрации препарата может произойти насыщение связей с белками, и тогда доля связанного препарата по отношению к общему его количеству начинает падать. Однако обычно концентрация свободного препарата слишком мала по сравнению с количеством вакантных связей, что препятствует существенному уменьшению связывания. Например, отношение связанной и свободной форм фентанила остается постоянным в пределах широких колебаний концентрации. Только при очень высокой концентрации (после болюсной инъекции до достижения полного смешивания) или при конкуренции нескольких препаратов за одни и те же вакантные связи отношение связывания уменьшается.
Связанная с белком форма препарата не способна диффундировать в ткани и, следовательно, давать фармакологический эффект. Это может произойти только после диссоциации в свободную форму. Отношение связывания может оказывать выраженное влияние на скорость перераспределения препарата, так как проникновение последнего через мембрану почти всегда пропорционально концентрации свободной формы. Например, лидокаин (30% свободной формы) проникает через мембрану несравненно легче, чем диазепам (3% свободной формы).
Связывание препарата приводит к снижению его концентрации в водной фазе плазмы, что может облегчить абсорбцию. Степень связывания значительно влияет также на скорость метаболизма препарата или его элиминацию. Так, связывание тубокурарина с белками плазмы замедляет распределение препарата во внеклеточной жидкости и задерживает его экскрецию с мочой и желчью.
Большая растворимость летучих анестетиков в плазме и цельной крови, чем это можно было бы объяснить их растворимостью в воде и жирах, обусловлена связыванием этих препаратов. Предполагается также, что связывание анестетиков белками играет важную роль в механизме общей анестезии на молекулярном уровне. Связывание фармакологических препаратов может происходить не только в плазме, но и в других тканях: жировой, соединительной, костной, эритроцитах.
Для того чтобы наступил фармакологический эффект, лекарственное средство должно попасть в ткань или орган в достаточной концентрации. В данном случае речь идет о концентрации в так называемой биофазе, т.е. концентрации медикамента в непосредственной близости от рецептора. В большинстве случаев препарат вводят в области, весьма удаленные от места действия, к которому он должен быть доставлен кровью, а затем через межклеточное пространство. На этом пути лекарственное средство должно пройти через множество барьеров, большинство из которых составляют мембраны.
Практически все мембраны в организме имеют общее строение. Они состоят из биомолекулярного слоя липидов, покрытого с обеих сторон одномолекулярным слоем белков.
Мембраны не образуют полностью непрерывного слоя: в них имеется множество пор радиусом около 9,4 нм.
Существуют четыре основных механизма транспорта лекарственного средства через мембраны:
жировая диффузия, при которой фармакологический препарат растворяется в жировом слое мембраны и диффундирует в соответствии с градиентом концентрации в водную фазу по другую сторону мембраны;
водная диффузия, при которой жирорастворимые гидрофильные вещества могут проходить через поры мембраны, чему способствует разница гидростатического и осмотического давления по обе стороны мембраны, обеспечивающая ток воды, которая и "протаскивает" лекарственный препарат через поры. Этот вид транспорта ограничен размерами молекулы препарата (у большинства лекарственных средств радиус молекулы значительно превышает 0,4 нм). Клетки капиллярного эндотелия имеют каналы радиусом до 4 нм, которые способны пропускать из плазмы в межклеточную жидкость или гломерулярный фильтрат даже молекулы альбумина;
активный транспорт, обеспечивающий движение вещества против концентрационного или электрохимического градиента (примером активного транспорта является широко распространенная в организме "натриевая помпа");
фагоцитоз и пиноцитоз, которые могут обеспечить транспорт лекарственных веществ (с высокой молекулярной массой или существующих в форме молекулярных агрегатов) в виде поглощенных клетками небольших капелек.
На транспорт лекарственных препаратов влияет ряд факторов. Укажем основные из них. Чтобы проникнуть через мембрану, медикамент должен прежде всего раствориться в воде и в виде водного раствора войти в контакт с мембраной. Следовательно, скорость абсорбции препарата зависит от его растворимости в воде. Вступая в контакт с мембраной, многие препараты растворяются в жировой составляющей мембраны. Жировая диффузия зависит от разделительного коэффициента растворимости препарата между водной и жировой фазами мембраны. Чем выше этот коэффициент, тем скорее абсорбируется вещество.
Большинство фармакологических средств являются слабыми электролитами и содержат кислые или основные группы (или обе разновидности). В водном растворе они образуют ионы. Степень ионизации зависит от константы диссоциации. Поскольку клеточная мембрана почти непроницаема для ионизированной формы любого лекарственного соединения, становится понятной зависимость фармакологического эффекта от зтого показателя, о чем мы уже говорили. Степень ионизации влияет не только на абсорбцию, но и на экскрецию почками.
Заслуживает упоминания прохождение лекарственного препарата через мембраны, обладающие особыми свойствами. Мембраны мозговых капилляров напоминают липидные мембраны клеток и значительно менее проницаемы, чем у обычных капилляров. Этим и объясняется существование гематоэнцефалического барьера. Проникновение лекарственного препарата через барьер зависит от степени ионизации, растворимости в жирах, связывания по обе стороны барьера. Такие высокоионизированные препараты, как тубокурарин и гексаметоний, вообще не могут пересечь эту границу. Природное соединение - физостигмин по химическому строению является третичным амином и легко проникает в мозг. Прозерин, близкий по действию к физостигмину, будучи четвертичным амином и высокоионизированным при нормальном рН плазмы соединением, не оказывает действия на ЦНС.
Проникновение в мозг неионизированных молекул зависит от растворимости в жирах. Тиопентал-натрий, значительная часть которого находится в плазме в неионизированной форме, будучи хорошо растворимым в жирах, легко проникает в мозг. Другой барбитурат этаминал-натрий (нембутал) из-за низкой растворимости в жире проникает в мозг медленно.
Другой пример особых свойств - плацента, которую можно рассматривать как своеобразную липидную мембрану. Прохождение медикаментов через нее зависит главным образом от растворимости в жирах. Помимо растворимости в жирах и степени ионизации, на преодоление плацентарного барьера влияют степень связывания лекарственного соединения с белками плазмы и интенсивность плацентарного кровотока. Такие высокоионизированные препараты, обладающие к тому же низкой растворимостью в жире, как тубокурарин и дитилин, проходят через плаценту крайне медленно. Для ингаляционных анестетиков и тиопентал-натрия быстро устанавливается равновесие между кровью матери и плода. Морфин и другие наркотические анальгетики также легко попадают в кровь плода, что имеет важное значение для понимания путей профилактики депрессии новорожденных непосредственно после родов.
Фармакокинетика средств, применяемых в анестезиологииПомимо перечисленных факторов, влияющих на распределение и транспорт фармакологических средств, рассмотрим ряд понятий, используемых при оценке кинетики лекарственных препаратов.
Реакция (или процесс) первого порядка характеризует скорость изменения содержания препарата, пропорциональную его концентрации. Это означает, что чем выше концентрация лекарственного средства в данном секторе, тем большее его количество покидает сектор за единицу времени, и наоборот. Так ведет себя большинство медикаментов в обычной терапевтической концентрации (например, морфин, тиопентал-натрий).
Реакция (или процесс) нулевого порядка происходит с постоянной скоростью независимо от концентрации препарата, что обеспечивает элиминацию из сектора (или организма) постоянного количества фармакологического средства за единицу времени (примером может служить алкоголь). В некоторых случаях при введении слишком высоких доз препарата возможен переход от процесса первого порядка к нулевому. При этом элиминация замедляется и при продолжении поступления таких доз может быть достигнут токсический уровень.
Проникновение препаратов через биологические мембраны может происходить в виде процесса как первого, так и нулевого порядка, хотя чаще действует первый механизм.
Устойчивое состояние представляет собой динамическое равновесие, при котором поступление лекарственного препарата равно его выделению и содержание в секторе (или организме) остается постоянным.
Время "полужизни" (период, или время, полувыведения - Т1/2) - время, требующееся для снижения концентрации лекарственного препарата в плазме на 50% после завершения периода абсорбции. Этот показатель имеет значение только для фармакологических средств, для которых характерен кинетический процесс первого порядка, поскольку при процессе нулевого порядка элиминация постоянного количества медикамента происходит независимо от концентрации.
Концентрация в крови препаратов, вводимых внутривенно болюсно и подчиняющихся реакции первого порядка, вначале падает очень быстро. Это распределительная фаза, или а-фаза (Т1/2а), которая имеет значение только для небольшого числа препаратов. Тем не менее она представляет интерес для анестезиологов, часто производящих внутривенные болюсные инъекции (например, барбитуратов). Затем концентрация препарата падает более медленно. Это фаза элиминации - бета-фаза (Т1/2бета), в которой препарат окончательно выводится из организма. Некоторые авторы именно эту фазу определяют как "полужизнь" фармакологического средства. Следует иметь в виду, что период полувыведения отнюдь не идентичен прекращению на 50% фармакологического действия медикамента, хотя в целом длительность эффекта, безусловно, находится в прямой зависимости от периода полувыведения.
Условный ("кажущийся") объем распределения (Уо) характеризует зависимость между количеством препарата в организме и его концентрацией в плазме после абсорбции и распределения: VD = Xp/C, где XD - доза препарата; С - концентрация в плазме после полного распределения препарата в организме и достижения равновесия.
Следует подчеркнуть, что это полностью условная гипотетическая величина, предполагающая равномерность распределения лекарственного соединения по всем секторам организма и отражающая реальный анатомический объем. VD зависит главным образом от свойств медикамента, а не от истинной величины тела и может варьировать в широких границах. Большой объем Vd свидетельствует о широком распределении препарата или потреблении его тканями. Условный характер этого показателя и многочисленные допущения приводят к тому, что для некоторых препаратов VD может превышать истинный объем тела, указывая на то, что в некоторых тканях концентрация значительно выше, чем в крови. Наоборот, очень низкий Vd означает, что значительная часть препарата осталась в крови, а потребление его тканями невелико.
Элиминация (процесс удаления лекарственного препарата из организма) представляет собой сумму двух составляющих - метаболических преобразований и экскреции соединения. Превалирование того или иного компонента или их одновременное сочетание зависит от химических свойств соединений. Часть препаратов выделяется из организма неизмененными, другие должны подвергнуться процессу метаболизма до образования промежуточных продуктов, которые затем выделяются из организма.
На первый взгляд, клиницисту безразлично, каким путем и как быстро подвергается метаболическим превращениям фармакологическое соединение. Однако это не так. Понимание механизма и последствий метаболической трансформации лекарственных средств важно не только с точки зрения фармакокинетики, но и с позиций фармакодинамики:
образующиеся в процессе метаболизма промежуточные продукты могут обладать фармакологической активностью (например, образующийся при распаде дитилина сукцинилмонохолин способен вызывать релаксацию);
метаболиты могут давать токсический эффект (в частности, продукты метаболизма метоксифлурана способны вызывать поражение почек);
путем торможения метаболизма лекарственного препарата можно продлить его действие (так, применение антихолинэстеразных средств увеличивает длительность действия дитилина);
интенсификация метаболизма медикамента укорачивает длительность его действия (например, барбитураты способны уменьшать продолжительность эффекта непрямых антикоагулянтов). Два последних примера иллюстрируют практическое значение не только метаболизма медикаментов, но и взаимодействия лекарственных препаратов, к чему мы еще вернемся.
Следует отметить, что одним из главных путей зкскреции лекарственных средств является их выделение почками. Растворимые в жирах вещества легко проходят клубочковый аппарат и поступают в канальцы, где столь же легко реабсорбируются через липидные мембраны тубулярных клеток. Создается замкнутый цикл, выход из которого заключается в метаболических превращениях в водорастворимые (и менее липофильные) соединения.
В процессе метаболизма лекарственные средства подвергаются реакциям двух видов - так называемым функционализации и сопряжению (конъюгации). "Функционализация" заключается во введении, раскрытии или модификации специфических химических групп и осуществляется с помощью окисления, восстановления или гидролиза. "Сопряжение" подразумевает соединение самого лекарственного препарата или его главного метаболита, образующегося при "функционализации", с такими эндогенными соединениями, как уксусная, глициновая, сернистая или глюкуроновая кислоты. Получающиеся при этом вещества обычно (хотя и не всегда) обладают выраженной растворимостью в воде.
Метаболические преобразования происходят главным образом в печени, хотя могут протекать практически во всех тканях и клетках, особенно в легких, кишечнике, крови. Ферменты, осуществляющие окисление, восстановление, гидролиз или образование соединений с кислотами, располагаются в эндоплазматической сети, пронизывающей клетки и относящейся к микросомальному аппарату. Для метаболических превращений некоторых лекарственных средств требуется один из этих процессов, однако большинство нуждается по меньшей мере в двух.
Микросомальная ферментная система окисления фармакологических средств неспецифична и многофункциональна, что создает условия для биотрансформации практически всех лекарственных препаратов. Ее каталитические компоненты включают флавопротеиновые редуктазы, фосфолипиды и гемопротеины с NADPH и молекулярным кислородом в качестве кофакторов. Цитохром бета-450 служит терминальной оксидазой в сложной цепи реакций и принимает непосредственное участие в связывании медикамента с микросомами.
Другие реакции - восстановление, отсоединение радикала (например, диметилирование, деаминирование и др.), разрушение кольца ароматического соединения - осуществляются также с помощью микросомальных ферментов, хотя и реже, чем окисление. Довольно частой реакцией является гидролиз эфирных связей (примером служит гидролиз, вызываемый холинэстеразой). В гидролизе принимают участие также микросомальные ферменты.
Наконец, большую группу реакций составляет "сопряжение", при котором лекарственный препарат или продукты его метаболизма соединяются с эндогенными веществами (в их число входят производные обмена углеводов и аминокислоты). Укажем наиболее важные реакции "сопряжения": метилирование, при котором донором метила служит метионин (например, метилирование катехоламинов), ацетилирование (в частности, сульфаниламидов), образование производных глюкуроновой кислоты (довольно распространенная реакция, участвующая в метаболизме таких препаратов, как морфин, стероидные гормоны и др.), соединение с глицином, глутамином и др.
Активность метаболических преобразований лекарственных препаратов в организме зависит от множества факторов. Среди них наиболее важными являются особенности метаболизма, генетически присущие данному лицу, возраст больного, эффект внешних воздействий (оперативное вмешательство, анестезия), действие различных заболеваний, в особенности болезней печени и почек, и многие другие.
Вторая составляющая элиминации - экскреция, которая происходит через почки, легкие, кишечник. Лекарственные препараты могут выделяться в неизмененном виде или после метаболических превращений.
Выделение лекарственного средства с мочой определяется тремя факторами - гломерулярной фильтрацией, тубулярной реабсорбцией и тубулярной секрецией. Гломерулярная фильтрация зависит от молекулярной массы и концентрации свободного препарата в плазме. Через гломерулярные капилляры могут проходить практически все растворы. Однако медикаменты, связанные с белками плазмы, остаются в крови. В целом экскреция препаратов через почки зависит от растворимости в жирах, причем соединения с большим коэффициентом жир/вода и в неионизированной форме легко реабсорбируются из гломерулярного фильтрата через тубулярный эпителий. Ионы не способны переходить через эту границу. Лекарственные средства выделяются также в проксимальных канальцах. Здесь протекает активный транспорт органических кислот и оснований в виде ионизированных молекул. Фармакологические препараты могут попадать в мочу и путем пассивной диффузии через тубулярный эпителий.
Физико-химические факторы, влияющие на абсорбцию лекарств из желудочно-кишечного тракта (растворимость в воде, коэффициент жир/вода), воздействуют также на элиминацию этих соединений или их метаболитов в кишечник и дальнейшее удаление с калом. Кроме того, в качестве специализированного транспортного механизма экскреции лекарственных препаратов через желудочно-кишечный тракт действует желчная система. Этим путем выделяются главным образом препараты с высокой молекулярной массой, подвергающиеся метаболическим превращениям в печени. Существует также механизм активного транспорта кислот и оснований из крови в желчь, который напоминает механизмы, действующие в почках.
Поскольку элиминацию можно рассматривать как очищение, самой важной ее количественной характеристикой служит клиренс плазмы - скорость, с которой происходит элиминация лекарственного средства на единицу его концентрации в плазме. Клиренс служит также мерой эффективности элиминации и измеряется в литрах в час или в миллилитрах в минуту. Клиренс обратно пропорционален Т1/2 (чем меньше Т1/2, тем выше клиренс, и наоборот) и прямо пропорционален Vd (при любом Т1/2, чем больше объем распределения, тем больше клиренс).
Если лекарственное соединение не реабсорбируется (или не секретируется) в канальцах, его клиренс равен скорости гломерулярной фильтрации (130 мл/мин). Липофильные препараты - слабые кислоты и основания - мало ионизированы и подвергаются интенсивной реабсорбции. В таких случаях почечный клиренс всегда меньше скорости гломерулярной фильтрации. Если препарат выделяется путем активного транспорта (секреции) через канальцы, то клиренс соответствует почечному плазмотоку независимо от гломерулярной фильтрации. Чаще всего (за исключением препаратов, секретируемых в канальцах) почечный клиренс происходит по типу реакции первого порядка.
Печеночный клиренс - это объем крови, протекающей через печень за единицу времени, из которого полностью удаляется лекарственное средство. Таким образом, печеночный клиренс является производным печеночного кровотока и способности печени экстрагировать и метаболизировать лекарственный препарат.
Зависимость печеночного клиренса от печеночного кровотока, степени экстракции и метаболической активности печени иллюстрируют следующие примеры. Если печеночный кровоток уменьшается без каких-либо изменений метаболической активности, то препарат с очень высоким печеночным клиренсом удаляется намного медленнее, а удаление препарата с малым печеночным клиренсом почти не изменяется. Наоборот, если способность печени метаболизировать медикамент резко возрастает, то увеличение клиренса более выражено по отношению к лекарствам с исходно низким метаболизмом, чем к препаратам, которые исходно подвергаются интенсивной печеночной экскреции.
Говоря о печеночном клиренсе, следует упомянуть о так называемом эффекте первого пассажа. Это понятие характеризует потребление и элиминацию препарата в печени во время первого поступления его (пассажа) в кровоток при приеме внутрь, введении интраперитонеально или в сосуды системы портальной вены. В результате первого пассажа значительная часть принятой дозы препарата не достигает общего кровотока. Этот эффект имеет для анестезиологии и реаниматологии меньшее значение, чем общая концепция печеночного клиренса, поскольку в анестезиологической практике упомянутые пути введения используют сравнительно мало.
Завершая рассмотрение вопросов общей фармакокинетики, нужно отметить, что для характеристики и изучения в целом поведения лекарственных препаратов в организме предложены различные модели, поддающиеся графическому и математическому описанию. Они варьируют по сложности. Наиболее простая, односекторная модель предполагает, что лекарственный препарат распределяется мгновенно и равномерно во всех жидкостях и тканях организма. Привлекая простотой, эта модель не способна точно отразить истинное поведение фармакологического средства в организме во времени. Двухсекторная модель подразумевает, что медикамент распределяется между центральными и периферическими секторами. Эту модель используют чаще, и она во многом удовлетворительно "описывает" кинетическую судьбу препарата. Однако для некоторых лекарственных соединений требуется более сложный анализ, что заставляет использовать трехсекторную и даже многосекторные модели.
Под центральными секторами обычно понимают кровь и те органы, которые получают большую часть сердечного выброса (мозг, сердце, печень, почки) Однако эти секторы являются абстракцией и не "описывают" истинной кинетики процесса в определенном органе. Периферические секторы обычно обозначают ткани с ограниченным объемом перфузии крови (мышцы, скелет, жировая и соединительная ткань).
Применяемые в фармакокинетике модели многие клиницисты рассматривают как экзотические математические игры, ничего или мало дающие практической медицине. Это, конечно, не так. Более того, используемые модели открывают путь к пониманию, а в некоторых случаях и к предсказанию зависимости эффекта препарата от его кинетики. Тем не менее, как и вообще в математическом моделировании, фармакокинетические модели имеют ограничения. В связи с этим за последние годы наметилась тенденция к развитию так называемой физиологической фармакокинетики, которую отличает от классической использование не абстрактных, а конкретных величин, таких как размеры органа, объем кровотока в нем и т.д.
Список литературы
1. Закусов В.В., Комиссаров И.В., Синюхин В.Н. Общая фармакология // Клиническая фармакология / Под ред. В.В. Закусова. - М., 1978. - С.22-63.
2. Соловьев В.Н., Фарсов А.А., Фишман В.М. Фармакокинетика. - М.: Медицина, 1980.
3. Холодов Л.Е., Яковлев В.П. Клиническая фармакокинетика: Руководство. - М.: Медицина, 1985.
4. Gibaldi М., Levy G. Pharmacokinetics in clinical practice, a. Applications // J. A. M. A. - 1976. - Vol.235, N 18. - P. 1987-1992.
5. Hug С.С. Pharmacokinetics and dynamics of narcotic analgesics // Pharmacokinetics of anaesthesia / Ed. C. Prys-Roberts, С.С. Hug. - Oxford, 1983. - P.187-234.
6. Hull C. J. General principles of pharmacokinetics // Pharmacokinetics of anaesthesia / Ed. C. Prys-Roberts, C. C. Hug. - Oxford, 1983. - P.1-24.
Похожие работы
... (Г1/2бета менее 5 ч) является завоевавший популярность в западных странах мидазолам. Наркотические анальгетики занимают важное место в арсенале средств, используемых в анестезиологии, что, естественно, повышает интерес к фармакокинетике этих препаратов. Изучение их действия осложняется рядом обстоятельств (многообразие путей введения, несовершенство методов определения концентрации анальгетиков ...
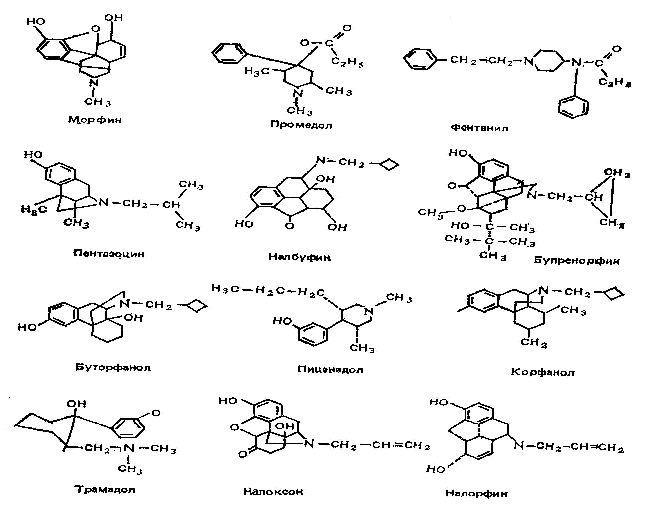
... в клинической практике. Одним из таких препаратов является просидол. Изучаемый новый наркотический анальгетик просидол (производное оксипиперидина), разработан в Институте химических наук им. А.Б. Бектурова МН АН РК, под руководством члена-корреспондента АН РК д.х.н. профессора Пралиева К.Д. До начала проведения клинических исследований были получены данные от создателей препарата о его ...
... характерной чертой которых является глобальное дезорганизующее действие на ЦНС с развитием потери сознания и снижения реакций на внешние раздражители. Другие средства внутривенной общей анестезии с избирательным диапазоном центрального действия (наркотические анальгетики, транквилизаторы, нейролептики) рассматриваются в разделе, посвященном специальным методам комбинированной общей анестезии. ...
... психотропных средств применение для премедикации наркотических анальгетиков мало оправдано, исключая больных с болевым синдромом. Холиноблокирующие (холинолитические, антихолинергические) средства. Для премедикации применяются две группы этих веществ – периферического и центрального (преимущественно м-холинолитического) действия. Холинолитические препараты периферического действия (атропин, ...
0 комментариев