Электрогравиметрия (ЭГМ) является разновидностью гравиметрии. Особенность ЭГМ заключается в осаждении определяемого элемента путем электролиза на предварительно взвешенном электроде. О массе элемента в растворе судят по увеличению массы электрода после электролиза.
ЭГМ применяют для определения металлов из растворов, в которых они присутствуют в виде ионов.
При электролизе катионы перемещаются к катоду, выделяясь на нем в виде металлов. Только очень немногие металлы осаждаются на аноде. К ним относятся, например, Mn и Pb, окисляющиеся в процессе электролиза до MnO2 и PbO2.
ЭГМ применяют для определения металлов, дающих плотные осадки на электроде, не осыпающиеся при промывании, высушивании и взвешивании. Кроме того, ЭГМ применяют только в тех случаях, когда осаждение определяемого металла не сопровождается соосаждением других металлов или примесей.
Электроды, применяемые в ЭГМ, должны отвечать следующим требованиям:
1) быть химически инертными;
2) хорошо удерживать образующиеся осадки;
3) иметь возможно меньшую массу и возможно большую поверхность;
4) не препятствовать перемешиванию раствора.
Всем этим требованиям в наибольшей степени удовлетворяют платиновые сетчатые электроды. Анодом, в большинстве случаев, служит платиновая проволока, согнутая в спираль.
Для проведения ЭГМ два платиновых электрода погружают в стакан с анализируемым раствором, подсоединяют электроды к внешнему источнику тока и проводят электролиз. При прохождении тока через раствор электролита происходят процессы восстановления и окисления соответствующих веществ на электродах. Связь между количествами веществ, участвующих в электродных процессах, и количеством электричества Q (Q = It) через цепь за время электролиза t при токе I устанавливается двумя законами Фарадея:
1) масса вещества, выделившаяся при электролизе, пропорциональна количеству электричества, прошедшего через раствор;
2) при прохождении через раствор одного и того же количества электричества на электродах выделяется одно и то же количество вещества эквивалента.
Математически оба закона можно представить формулой
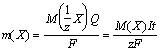 ,
,
где m(X) - масса вещества X, выделившегося при электролизе;
M(1/z X) и M(X) - молярная масса эквивалента и молярная масса вещества X, соответственно;
z - число эквивалентности;
F - число Фарадея, равное количеству электричества (96500 Кл), которое требуется для выделения 1 моль эквивалентов вещества.
Формула позволяет решать различные задачи, связанные с электролизом. Например, вычислить продолжительность при заданной силе тока для выделения определенной массы вещества. На практике электролиз требует больше времени, чем это следует из формулы. Это связано с побочными реакциями, обычно сопровождающими главные. Поэтому КПД тока, иначе называемый выходом по току, почти всегда ниже 100%.
Выход по току может быть определен как отношения массы вещества m, реально полученного при электролизе, к массе вещества, которая могла бы получиться в соответствии с законом Фарадея m0, если бы количество электричества не расходовалось на побочные процессы:
![]() .
.
При прохождении через раствор электрического тока на электродах выделяются продукты электролиза, что приводит к возникновению в системе ЭДС обратной внешней ЭДС источника тока. Это явление называется электрохимической поляризацией, а возникающая обратная ЭДС - ЭДС поляризации. Ее можно заметно уменьшить, прибавляя так называемые деполяризаторы, т.е. вещества, разрежающиеся прежде, чем ионы, которые разрежались бы в их отсутствие.
Таким образом, чтобы электролиз мог происходить, необходимо приложить к электродам напряжение, превышающее ЭДС поляризации. Наименьшее напряжение, которое необходимо приложить к электродам для того, чтобы вызвать непрерывный электролиз данного электролита, называется его напряжением разложения Ер. Ер должно быть больше ЭДС гальванического элемента Е (Е = Еа - Ек) на величину перенапряжения Ер = Е + = (Еа+ a) - (Ек - k), где Еа и Ек - равновесные потенциалы анода и катода, а а и к - перенапряжения на аноде и катоде.
Величина перенапряжения зависит от:
1) плотности тока j = I/S (где S - площадь поверхности электрода). Чем больше j, тем больше ;
2) состояния поверхности электрода: на гладком электроде больше, чем на шершавом, так как при одинаковой силе тока приходящаяся на единицу поверхности плотность тока больше;
3) температуры: повышение температуры уменьшает ;
4) природы электрода и различных примесей в растворе.
При электролизе нужно учитывать силу тока в цепи. Чем больше I, тем больше j и тем больше в единицу времени на поверхности электрода выделится определяемого металла. Следовательно, тем быстрее закончится электролиз и анализ в целом.
Однако при слишком большой j осадок получается рыхлым (губчатым), непрочно связанным с электродом. Причина этого в том, что при слишком большой j скорость разрядки ионов определяемого металла становится больше скорости их подвода к электроду. Поэтому раствор около катода начинает настолько обедняться ионами, что на катоде начинает восстанавливаться водород, пузырьки которого разрыхляют осадок. Введение комплексообразующих компонентов предотвращает выделение водорода и способствует получению прочных однородных осадков металлов.
Многие металлы, например Zn, Sn, Pb, при низких плотностях тока выделяются в виде непрочного слоя. Предполагается, что причина этого - присутствие в электролите растворенного кислорода и примесей окислителя.
Условия электролиза должны быть выбраны так, чтобы происходило выделение только одного металла, а не их смеси, и чтобы выход по току составлял 100%.
После электролиза электроды промывают несколько раз дистиллированной водой, не отключая электроды от источника тока, затем сушат и точно взвешивают. По разности масс электродов, без осадка и с ним, находят массу определяемого вещества в растворе.
Внутренний электролиз ЭГМ можно выполнить в накоротко замкнутом гальваническом элементе. При этом не требуется внешнего источника тока, так как осадок выделяется за счет энергии гальванического элемента. Такой вариант ЭГМ называют внутренним электролизом.
Например, ионы Cu2+ будут количественно выделяться из раствора на платиновом сетчатом катоде, если этот электрод соединить с цинковым анодом. В полученном таким образом гальваническом элементе Zn, имеющий меньший окислительный потенциал, переходит в раствор, отдавая электроны:
Zn - 2![]() Zn2+.
Zn2+.
Высвобождающиеся при этом электроны переходят по проводу к платиновому электроду, который передает их Cu2+-ионам, восстанавливая их до металлической меди, оседающей на платиновой сетке:
Cu2+ + 2![]() Cu .
Cu .
Вместо Zn можно употреблять и другие металлы (Al, Fe, Pb и др.), имеющие потенциал, более отрицательный, чем у выделяемого металла.
Слабый и очень равномерный ток при внутреннем электролизе дает возможность выделять даже чрезвычайно малые количества металла, который покрывает катод очень ровным и плотным слоем.
Главной опасностью при внутреннем электролизе является цементация, т.е. разряжение части ионов определяемого металла непосредственно на самой анодной пластинке. Для предотвращения цементации катод отделяют от анода перегородкой (диафрагмой), чаще всего из коллодия, или покрывают анод полупроницаемой пленкой коллодия.
Ю.Ю. Лурье установлено, что при малых количествах (не более 20 мг) определяемого металла диафрагму можно не применять. Во избежание цементации необходимо, чтобы течение тока во время электролиза ни на минуту не прерывалось, и электроды не касались друг друга. Материал анода должен не сдержать примесей, поверхность его должна быть невелика и хорошо отшлифована. Результат анализа получают по привесу платиновой сетки по окончании электролиза. Можно также осевший металл растворить и окончить определение фотометрическим или другим методом.
Кулонометрия - это метод, основанный на измерении количества электричества, израсходованного на электропревращении (восстановлении или окислении) определяемого вещества. Если протекает какая-либо электрохимическая реакция MeZ+ + Ze - Me0, то по количеству электричества, израсходованного на эту реакцию, числу электронов, принимающих в ней участие, можно определить содержание вещества по закону Фарадея.
Необходимыми условиями применения метода является, как и в ЭГМ, 100% -й выход по току и отсутствие конкурирующих реакций.
Электролиз в кулонометрической ячейке можно проводить при постоянной силе тока (гальваностатическая кулонометрия), либо при постоянном потенциале (потенциостатическая кулонометрия). В свою очередь по методике выполнения различают прямую и косвенную кулонометрию (кулонометрическое титрование). Прямые определения обычно проводят при постоянном потенциале рабочего электрода, а косвенные - при постоянной силе тока.
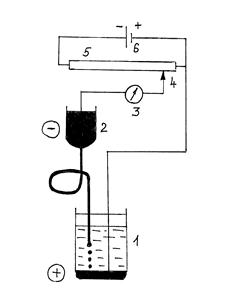
Схема установки для прямой кулонометрии (рис.2.12.1) включает кулонометрическую ячейку 1, источник постоянного напряжения (потенциостат) 2, вольтметр 3. Электролиз ведут до тех пор, пока не закончится электропревращение определяемого вещества на электроде, то есть сила тока, контролируемая амперметром 4, не уменьшится до незначительной величины.
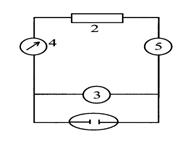
Рис. 1. Схема установки для прямой кулонометрии.
Для измерения количества прошед-шего электричества может служить специальное измерительное устройство - кулонометр. Принцип его действия основан на том, что через этот последовательно включенный прибор протекает такой же ток, какой проходит через анализируемый
раствор и, следовательно, за то же время, то же количество электричества. В кулонометре со 100% -м выходом протекает хорошо известная электрохимическая реакция, и измерение Qх сводится к определению Qст., полученного в результате этого процесса. В зависимости от способа измерения объема, массы вещества различают электрогравиметрические титрационные и другие кулонометры. В газовых кулонометрах определяется масса газа, выделившегося в результате электрохимического процесса. В электрогравиметрических - масса вещества; напри мер, масса меди, выделившейся при электролизе CuSO4 в медных куло нометрах, масса Ag при электролизе AgNO3в серебряных кулонометрах.
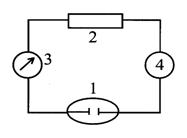
Рис. 2. Схема установки кулонометрического титрования.
При проведении кулонометрического титрования используют уста-новку, включающую ячейку 1, источник постоянного тока (гальваностат) 2, амперметр 3. Время электролиза могут измерять включенным в схему электрохронометром 4 или секундомером (рис.2.12.2).
В основе прямой кулонометрии лежит непосредственное электропревращение определяемого вещества на электроде, поэтому метод пригоден только для определения электроактивных веществ, т.е. веществ, способных окисляться или восстанавливаться на электроде.
Прямые определения проводят при предварительно подобранном постоянном потенциале рабочего электрода, обеспечивающем устранение конкурирующих реакций и 100% -ю эффективность по току. Момент окончания электродной реакции определяют, используя зависимость I = f(t), которая в перемешиваемом растворе выражается уравнением It = I010-Kt, It - сила тока в момент времени t, I0 - в начальный момент; K - константа, зависящая от площади поверхности электрода S, коэффициента диффузии D объема раствора V и толщины диффузионного слоя : K = 0,43 DS/V. Из уравнения следует, что для полного восстановления или окисления определяемого вещества на электроде требуется бесконечно большее время, что практически неосуществимо. Поэтому электролиз ведут не до I = 0, как это требуется для 100% -ного выхода по току, а до достижения небольшого, не изменяющегося в течение времени тока, составляющего 0,01 - 0,001% I0. Для ускорения завершения электролиза используют электроды с большой площадью, малый объем раствора и перемешивание. При правильно выбранных условиях электролиза его время не превышает 0,5 ч.
Прямая кулонометрия - высокочувствительный и точный метод, позволяющий с помощью современных приборов определять до 10-9 г вещества, за время 103с с погрешностью не более 0,5%. Этот метод безэталонный и легко автоматизируемый.
В процессе кулонометрического титрования определяемое вещество реагирует с титрантом, получаемым в результате электрохимической реакции на электроде. Такой титрант называют электрогенерированным, а электрод, на котором его получают - генераторным. Такой титрант можно получить из растворителя, например, воды на катоде:
Н2О + ![]() ½Н2 + ОН - или аноде Н2О - 2
½Н2 + ОН - или аноде Н2О - 2![]() ½О2 + 2Н+.
½О2 + 2Н+.
Электрогенерированные ионы ОН - и Н+ можно использовать для титрования кислот или оснований.
Чаще титрант генерируют из специального вещества, вводимого в ячейку, обычно называемого вспомогательным реагентом (см. таблицу 2.12.1). Реакцию электролиза проводят при I = const.
Для обеспечения 100% выхода по току в ячейку вводят избыток вспомогательного реагента. Если титрант генерируется в количестве, эквивалентном содержанию определяемого вещества, то определив Q, затраченное на получение титранта, можно определить содержание определяемого вещества. Поэтому необходимо иметь надежный способ фиксирования конца титрования. Для этого можно применять химические индикаторы для визуального установления КТТ, так и инструментальные методы, например потенциометрию.
Достоинством кулонометрического титрования является то, что титрант не нужно готовить, хранить и стандартизировать, так как метод безэталонный, абсолютный и позволяет оценить количество определяемого вещества, а не его концентрацию.
Таблица 1.
| Титрант | Вспомо-гательный реагент | Реакция на электроде | Титруемые вещества | Тип титрования |
| OH - H+ | H2O H2O | 2H2O+2e2OH-+H2 2H2O-4eO2+4H+ | Основание кислота | Кислотно-основное |
| Ag+ | Ag-анод | AgAg++e | Cl-,Br-, I-, серосодержащие органические вещества | Осади-тельное |
| Mn2+ Br2 CuCl3 - Cl2 I2 | MnSO4 KBr CuCl2 KCl KI | Mn2+Mn3++e 2Br-Br2+2e Cu2++3Cl-+eCuCl3 - 2Cl-Cl2+2e 2I-I2+2e | Fe(II),H2C2O4 Sb(III), I, фено-лы Cr(VI), IO3 - I-,As(III), S2O32-, As(III) | Окисли-тельно-восста-новительное |
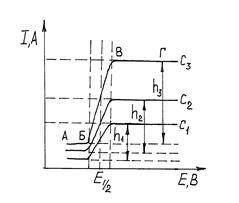
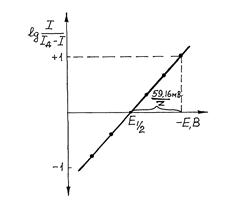
Вольтамперометрия основана на изучении поляризационных или вольтамперных кривых (кривых зависимости силы тока I от напряжения Е), которые получают в процессе электролиза раствора анализируемого вещества при постепенном повышении напряжения с одновременной фиксацией при этом силы тока. Электролиз проводят с использованием легкополяризуемого электрода с небольшой поверхностью, на котором происходит электровосстановление или электроокисление вещества.
Вольтамперометрию, связанную с использованием ртутного капающего электрода (РКЭ), называют полярографией. Ее открытие в 1922 г. принадлежит чешскому ученому Я. Гейровскому, который в 1959 г. получил за этот метод Нобелевскую премию. Характерной особенностью полярографического метода является применение электродов с разной площадью поверхности. Поверхность одного из электродов, называемого микроэлектродом, должна быть во много раз меньше поверхности другого электрода. В качестве микроэлектрода чаще всего применяют РКЭ, представляющий собой капилляр, из которого равномерно с определенной скоростью вытекают капли металлической ртути. Скорость прокапывания определяется высотой подвески емкости с ртутью, соединенной шлангом с капилляром. Второй электрод, поверхность которого во много раз больше поверхности микроэлектрода, служит электродом сравнения. В качестве него используют ртуть, налитую на дно электролитической ячейки, или насыщенный каломельный электрод. На эти электроды от внешнего источника напряжения подают плавно изменяющееся напряжение. Плотность тока (А/см2) на электроде сравнения, имеющего большую поверхность, ничтожно мала, поэтому потенциал его практически не изменяется, т.е. этот электрод не поляризуется. Плотность тока на РКЭ вследствие его малой поверхности высока. РКЭ изменяет свой равновесный потенциал, т.е. поляризуется. Реализацию метода осуществляют на приборах, называемых полярографами.
Похожие работы
... электропроводности растворов от концентрации тех или иных электролитов. Малая эффективность является существенным ограничением метода ПК. Более широко применяется косвенная кондуктометрия - кондуктометрическое титрование (KT). КТ основано на заметном изменении характера зависимости электропроводности раствора от количества добавляемого титранта вблизи точки эквивалентности вследствие изменения ...
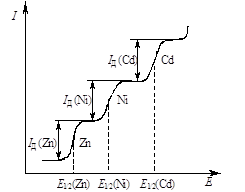
... тока. Электролиз проводят с использованием легкополяризуемого электрода с небольшой поверхностью, на котором происходит электровосстановление или электроокисление вещества. Вольтамперометрию, связанную с использованием ртутного капающего электрода (РКЭ), называют полярографией. Ее открытие в 1922 г. принадлежит чешскому ученому Я. Гейровскому, который в 1959 г. получил за этот метод Нобелевскую ...
... прямого проведения химического анализа, но могут применяться как вспомогательные в других методах анализа. Например, использоваться в титриметрии для регистрации конца титрования не с помощью химического цветопеременного индикатора, а по изменению потенциала, электрической проводимости тока и т.д. Теоретические основы ЭМА Электрод представляет собой систему, в простейшем случае состоящую ...
0 комментариев