Титрование с применением окислительно-восстановительной реакции. Характеристика окислительно-восстановительных реакций с помощью электродных потенциалов (возможность, направленность, влияние различных факторов). Каталитические, автокаталитические и сопряженные окислительно-восстановительные реакции.
Редокс-ТКТ и редокс-индикаторы, расчет индикаторной погрешности
Химические элементы, имеющие переменную степень окисления, могут быть количественно определены титриметрически с применением окислительно-восстановительной реакции (ОВР). Методы окислительно-восстановительного титрования называют оксред, или редоксиметрией (от латинского oxydatio - окисление и reductio - восстановление). По веществу титранта оксредметрию подразделяют на оксидиметрию и редуциометрию. В оксидиметрии в качестве вещества рабочего раствора используют окислители, а в редуциометрии - восстановители.
В зависимости от решаемой аналитической задачи в редоксиметрии используют прямое, обратное и заместительное титрования. Редокси-метрически могут быть количественно определены как неорганические, так и органические вещества. Например, восстановлением с помощью перманганата калия в сильнощелочной среде могут быть определены метанол, муравьиная, винная, лимонная, салициловая кислоты, а также глицерин, фенол, формальдегид и др.
Схематично ОВР, с учетом закона электронейтральности раствора, можно изобразить следующим образом:
OX1+z1e - Red1 z2
Red2-z2e - OX2 z1
z2OX1+z1Red2↔z2Red1+z1OX2
Здесь индексы 1 и 2 относятся к веществам 1и 2 в окисленной (OX1 и OX2) и восстановленной (Red1 и Red2) формах. В ходе ОВР вещество ОХ1 с большим сродством к электрону (окислитель) присоединяет электроны, понижает свою степень окисления, восстанавливается, а вещество Red2 с меньшим сродством к электрону (восстановитель) окисляется.
Окисленная и восстановленная формы реагирующих в ОВР веществ образуют окислительно-восстановительные (оксред-, редокс-) пары ОХ1/Red1 и OX2/Red2, а превращения типа OX+ze - Red называют оксред - (редокс) - переходами или окислительно-восстановительными полуреакциями. Если редокс-переходы обратимы, т.е. могут протекать при изменении условий как в одну, так и в другую сторону, то для количественной оценки редокс-свойств редокс-пар используют редокс - (окислительно-восстановительные) потенциалы. Редокс-понциал редокс-пары может быть электрохимически измерен. Для этого в раствор с компонентами редокс-пары (например, MnO4-/Mn2+) можно поместить платиновую пластинку (проволоку), не реагирующую с веществами редокс-пары, образовав таким образом редокс-электрод. Соединив платину редокс-электрода со стандартным водородным электродом, получим гальванический элемент, ЭДС которого можно измерить потенциометрически (см. гл.2.11). ЭДС гальванического элемента равна разности потенциалов электродов его составляющих. Поскольку потенциал стандартного водородного электрода равен нулю, то измеренная величина ЭДС равна величине потенциала редокс-электрода, т.е. редокс-потенциалу редокс-пары при данном соотношении концентраций её компонентов и прочих условиях измерения.
Зависимость редокс-потенциала E(OX/Red) от концентрации и температуры передается уравнением Нернста:
E(OX/Red) = E(OX/Red) +![]()
где E(OX/Red) - реальный или равновесный редокс-потенциал, В; E(OX/Red) - стандартный редокс-потенциал, равный равновесному при а(ОХ) = а(Red) = 1 моль/дм3;
R - универсальная газовая постоянная (8,31 Дж/Кмоль);
Т - абсолютная температура, K; F - число Фарадея (96500 Кл/моль);
z - число электронов, участвующих в редокс-переходе в ОХ+ze - dRed; a(OX) и a(Red) - активности соответственно окисленной и восстановленной форм вещества, моль/дм3.
При подстановке в уравнение Нернста значений R, F и T = 298 К, а также переходе к десятичному логарифму, получим
E(OX/Red) =E(OX/Red) + 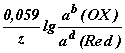 ,
,
так как а = γc, то
E(OX/Red) =E(OX/Red) + ![]() .
.
Обозначив первые два слагаемых в последнем уравнении E(1) (OX/Red), получим
E(OX/Red) =E(1) (OX/Red) + 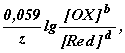
где E(1) (OX/Red) - формальный потенциал, характеризующий редокс-пару, у которой [OX] = [Red] =1 моль/дм3. E=E(1), если ионным взаимодействием между ОХ и Red можно пренебречь. Обычно это возможно при концентрациях ОХ и Red меньше 0,1 моль/дм3. Редокс-потенциал зависит также от кислотности среды, комплексообразования или осаждения одного из компонентов редокс-пары в процессе редокс-перехода. Чем больше концентрация ионов водорода в растворе, тем больше окислительная способность окисленной формы вещества редокс-пары и тем больше E(OX/Red).
Например, для редокс-перехода
MnO4-+5e-+8H+ Mn2++4H2O
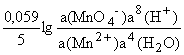
E(MnO4-/Mn2+) =E(MnO4Mn2+) +.
Для простоты расчетов а(Н2О) и а(Н+) обычно принимают равными единице.
Комплексообразование, как и образование малодиссоциирущего соединения, выводит закомплексованную или осажденную часть компонентов редокс-пары из участия в редокс-переходе. Из формулы Нернста следует, что комплексообразование или осаждение ОХ-формы уменьшает величину E(OX/Red), а Red-формы вещества - увеличивает.
Например, в отсутствие комплексообразования редокс-потенциал редокс-пары Fe3+/Fe2+ рассчитывался бы по формуле
E(Fe3+/Fe2+) = E(Fe3+/Fe2+) + ![]() .
.
Комплексование формы Fe3+ в присутствии, например, ЭДТУ (см. гл.1.10) в соединение FeY - приводит к изменению активности Fe3+ до величины 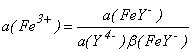 ,
,
где a (Y4-) - активность ЭДТУ; (FeY-) - константа устойчивости комплекса FeY-, т.е. константа равновесия реакции комплексообразования
Fe3++Y4 - FeY-.
После подстановки в формулу Нернста получим
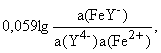
E(Fe3+/Fe2+) = E(FeY-/Fe2+) +
где E(FeY-/Fe2+) =E(Fe3+/Fe2+) - 0,059lg(FeY-), и если без комплексообразования E(Fe3+/Fe2+) = 0,77 В, то при комплексообразовании с ЭДТУ E(FeY-/Fe2+) = - 0,77 В (так как (FeY-) =1025,1).
Образование малорастворимых соединений при йодометрическом определении Cu2+ (осадка CuI и ионов Cu+ и I-) приводит к тому, что в уравнении
E(Cu2+/Cu+) = E(Cu2+/Cu+) + 0,059lg![]()
активность иона Cu+ становится равной
![]()
поэтому E(Cu2+/Cu+) = E(Cu2+/Cu+) + 0,059lg a(Cu2+) a(I-),
где E(Cu2+/CuI) = E(Cu2+/Cu+) - 0,059lgKs(CuI), и если E(Cu2+/Cu+) = 0,159 В, то E(Cu2+/CuI) = 0,865 В (так как Ks(CuI) =10-11,96).
При выборе вещества титранта в редоксиметрии проводят качественную и количественную оценку возможности (направленности) и полноты прохождения ОВР между титрантом и определяемым веществом.
Качественную оценку проводят путем сравнения табличных величин E(OX|Red) вещества титранта и определяемого вещества, приведенных в аналитических, химических и физико-химических справочниках. При этом руководствуются следующими правилами:
а) окисленная форма вещества редокс-пары с большим E играет роль окислителя по отношению к восстановленной форме вещества редокс-пары с меньшим E;
б) чем больше E, тем чётче выражена окислительная способность окисленной формы редокс-пары;
в) ОВР протекает в заданном направлении, если ЭДС= E(OX1/Red1) - E(OX2/Red2) > 0, причем чем больше ЭДС, тем интенсивней ОВР;
г) ОВР идут в сторону образования более слабых окислителей и восстановителей.
Количественной характеристикой направления и полноты протекания ОВР является её константа равновесия Кравн. Формулу для расчета Кравн можно вывести из условия установления состояния равновесия в редокс-системе, когда E(OX1/Red1) становится равным E(OX2/Red2). В этот момент
![]()
![]() .
.
После преобразования этого равенства получим

E(OX1/Red1) - E(OX2/Red2) = .
В этой формуле под знаком логорифма стоит выражение для Кравн., поэтому
lgKравн=![]() .
.
ОВР протекает в прямом направлении при Кравн>1 и тем полнее, чем больше Кравн. При Кравн ≥ 103 ОВР практически необратима.
Особенностью ОВР является сложный характер, определяемый многостадийностью с образованием промежуточных, часто нестойких и высокоактивных продуктов. Например, реакция окисления йодидионов пероксидом водорода, представляемая суммарным уравнением
2I-+H2O2+2H+ I2+2H2O, в действительности протекает в несколько стадий:
I-+H2O2 IO-+H2O
IO-+H+ HIO
HIO+I-+H+ I2+H2O
Скорость ОВР мала. Увеличить её можно повышением температуры до 100С или введением катализаторов. В качестве катализатора могут выступать Н+ - ионы. Катализаторы действуют на ОВР весьма специфично, ускоряя одни и не ускоряя другие. Например, окисление I - ионов ускоряет МnО4-, катализатором восстановления Ge4+ является OsO4, а MnO4 - OsO4 и KI.
Катализатором может быть продукт самой ОВР. Например, образующиеся ионы Mn2+ ускоряют реакцию
2MnO4+5C2O42-+16H+2Mn2++10CO2+8H2O.
Подобные реакции называют автокаталитическими.
Существенные осложнения в анализе могут вызвать образующиеся в ходе ОВР промежуточные высокоактивные соединения, способные вступать в побочные реакции. Типичным примером является перманганатометрическое определение Fe2+ в солянокислом растворе. В этом случае наблюдается повышенный расход KМnO4 по сравнению с титрованием в среде серной кислоты. Причиной является расход части количества KMnO4 на окисление Cl - ионов соляной кислоты до свободного Cl2:
2MnO4-+ 10Cl-+16H+2Mn2++5Cl2+8H2O,
причем в отсутствие ионов Fe2+ эта реакция не идет. Реакции подобного типа, не идущие одна без другой, Н.А. Шилов назвал сопряженными или индуцированными. При взаимодействии ионов MnO4 - и Fe2+ образуются неустойчивые соединения марганца в промежуточных степенях окисления: Mn(V), Mn(IV), Mn(III), которые, обладая высокой химической активностью, окисляют не только ионы Fe2+, но и Cl - ионы. Образовавшийся Cl2 частично улетучивается из раствора, что увеличивает расход KMnO4 на титрование Fe2+ - ионов.
Вещество, участвующее в обеих сопряженных реакциях, называется актором (в рассмотренном примере - MnO4-), вещество, реагирующее непосредственно с актором - индуктор (Fe2+), а реагирующее с актором только в присутствии индуктора - акцептор (Cl-).
Для предотвращения индуцированного окисления Cl - в присутствии Fe2+ рекомендуется при титровании раствором KМnO4 в титруемый раствор вводить MnSO4. Введенные ионы Mn2+ значительно легче окисляются неустойчивыми соединениями марганца до Mn7+, чем Cl - до Cl2, предотвращая газообразование хлора, поэтому взаимодействие ионов MnO4 и Fe2+ становится химически эквивалентным.
Перманганатометрия - безындикаторный метод. В ней окончание титрования устанавливают по порозовению титруемого раствора в КТТ при избытке раствора KМnO4, остальные редоксиметрические методы - индикаторные.
Правильный выбор редокс-индикаторов для регистрации КТТ редокс-титрования проводят по соответствующим ТКТ, которые строят на основе расчета редокс-потенциалов для различных моментов титрования. Точку “до начала титрования“ не рассчитывают, так как без добавления титранта к титруемому раствору ОВР не идет и редокс-система не образуется. Точки ТКТ “до ТЭ” рассчитывают по уравнению Нернста для той редокс-пары, в которою входит определяемое вещество, а “после ТЭ” - по уравнению для редокс-пары титранта. Потенциал в ТЭ вычисляют по формуле
Eэкв = ![]() .
.
Расчеты могут быть значительно упрощены, если в формулу Нернста вместо соотношения концентраций ввести степень оттитрования . (табл.1.9.1)
Таблица 1.9.1
| Участок ТКТ | Титруемое вещество | |
| восстановитель | окислитель | |
| До ТЭ После ТЭ | E=E1+ | E=E1+ |
Величина скачка титрования на редокс-ТКТ тем больше, чем больше разность потенциалов редокс-пар и концентрации анализируемого раствора и титранта. Принцип выбора редокс-индикатора по ТКТ тот же, что и в протолиметрии.
Изменение окраски редокс-индикаторов происходит под действием окислителя или восстановителя, сдвигающего равновесие между окисленной и восстановленной формами индикатора при изменении потенциала редокс-системы:
Ind(OX) +ze - Ind(Red).
Потенциал редокс-пары Ind(OX) /Ind(Red) может быть вычислен по уравнению Нернста:
E = E+![]() .
.
Учитывая, что изменение цвета раствора на глаз наиболее различимо, когда концентрация одной из форм индикатора в десять и более раз превышает концентрацию другой, т.е. при 1/10 [Ind(OX)] / [Ind(Red)] 10/1, после подстановки этого соотношения в формулу Нернста и преобразований получим интервал перехода редокс-индикатора в вольтах:
E=E![]() .
.
Для большинства редокс-индикаторов z в этой формуле равно двум.
Одним из наиболее известных и употребимых редокс-индикаторов является дифениламин
Под действием окислителей дифениламин необратимо переходит в бесцветный дифенилбензидин который затем обратимо окисляется до сине-фиолетового дифенилбензидинфиолетового
Eдифениламина = 0,76 0,029 В.
Возможные редокс-индикаторы и их характеристики приведены в аналитических и химических справочниках.
Несовпадение потенциала изменения окраски редокс-индикатора E(Ind) и потенциала титруемого раствора в ТЭ EТЭ приводит к индикаторной погрешности.
Например, при титровании с дифениламином (E(Ind) = 0,76 В) раствора ионов Fe2+ раствором K2Cr2O7 EТЭ=1,25 B. Поскольку E(Ind) < EТЭ, то часть ионов Fe2+ останется неоттитрованной, что приведет к отрицательной индикаторной погрешности.
Относительную индикаторную погрешность (Δ,%), равную молярной доле неоттитрованных ионов Fe2+, можно рассчитать следующим образом:
![]() ,%
,%
Для расчета Δ необходимо знать соотношение концентрации [Fe2+] от / [Fe2+] неот. в КТТ.
В КТТ [Fe2+] от= [Fe3+] КТТ, [Fe2+] неот= [Fe2+] КТТ, a EКТТ(Fe3+/Fe2+) = E(Ind), поэтому можно записать, что момент окончания титрования
![]()
EKTT(Fe3+/Fe2+) = E(Fe3+/Fe2+) + 0,059lg
откуда![]()
E(Ind) - E(Fe3+/Fe2+) + 0,059lg![]()
следовательно,
Поставим в эту формулу E(Ind) = 0,76 B для дифениламина и E(Fe3+/Fe2+) = 0,77 В, в результате получим [Fe3+] / [Fe2+] = 0,6768, а соответствующее этому ∆ = 59,63%, т.е. индикаторная погрешность при титровании с дифениламином недопустимо велика.
В общем случае относительную индикаторную погрешность при редоксиметрии восстановителей можно рассчитать по формулам (табл.1.9.2).
Таблица 1.9.2
| Причина погрешности | Формула расчета погрешности при титровании | |
| восстановителя | окислителя | |
| Недотитрование |
|
|
| Перетитрование |
|
|
Примечание: а = ![]() , zв-ва и Е0в-ва относятся к веществу (окислителю или восстановителю), которое в КТТ в избытке.
, zв-ва и Е0в-ва относятся к веществу (окислителю или восстановителю), которое в КТТ в избытке.
Похожие работы
... с увеличением концентраций реагирующих веществ, величины константы устойчивости комплексоната и температуры. Комплексонаты ЭДТА с ионами металлов - бесцветные cоединения, как и ЭДТА, поэтому ТЭ комплексонометрического титрования фиксируют с помощью индикаторов. Для этих целей можно использовать кислотно-основные индикаторы, оттитровывая в их присутствии щелочью ионы водорода, образовавшиеся при ...
... электропроводности растворов от концентрации тех или иных электролитов. Малая эффективность является существенным ограничением метода ПК. Более широко применяется косвенная кондуктометрия - кондуктометрическое титрование (KT). КТ основано на заметном изменении характера зависимости электропроводности раствора от количества добавляемого титранта вблизи точки эквивалентности вследствие изменения ...
... несколько капель индикатора, который обесцвечивается в точке эквивалентности. Обратимо изменяют свою окраску при проведении броматометрических определений n-этоксихризоидин, -нафтофлавон, хинолиновый желтый и др. Достоинства и недостатки броматометрического метода. Броматометрический метод отличается рядом достоинств по сравнению с другими методами. 1. Бромат-бромидные растворы можно ...
... у возводимого в степень числа. 4. В промежуточных результатах брать на одну десятичную цифру больше, чем по правилам округления, а для оценки порядка вычислений округлять все числа до первой значащей. Математическая обработка результатов анализа На любом из перечисленных этапов количественного анализа могут быть допущены и, как правило, допускаются погрешности, поэтому, чем меньшее число ...
0 комментариев