Результаты полярографических измерений иногда искажаются появлением так называемых полярографических максимумов, т.е. резким (в несколько десятков раз) превышением тока на отдельных участках вольтамперных кривых над предельным диффузионным током. Существует ряд причин возникновения этих максимумов. Полярографические максимумы первого рода (рис.2.14.1) возникают в разбавленных растворах и вызываются неравномерным распределением потенциала вдоль электрода, (капли ртути). Это приводит к различию значений избыточной поверхностной энергии на разных участках поверхности и отсюда к тангенциальным движениям жидкой поверхности ртути: сокращению участков с высокой избыточной энергией за счет расширения участков с низкой.
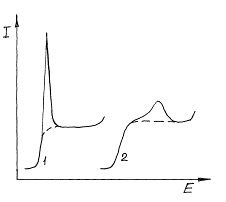
Рис. 1. Полярографические максимумы I рода (1) и II рода (2).
В результате этих движений резко увеличивается конвективная диффузия реагентов к поверхности. Эти максимумы образуются только на некотором удалении от потенциала нулевого заряда и обычно имеют вид узких высоких пиков тока.
Максимумы второго рода, наоборот, наблюдаются чаще в концентрированных растворах. Они охватывают более широкую область потенциалов, но меньше по высоте. Максимальная высота достигается вблизи точки нулевого заряда.
Причина их возникновения - тангенциальные движения поверхности ртутной капли, вызванные вытеканием ртути из капилляра (рис. 2): ртуть движется вниз по внутреннему объему капли и потом поднимается вдоль ее поверхности. Полярографические максимумы обоих родов могут быть устранены путем добавления поверхностно-активных органических веществ. Изменение адсорбции этих веществ, возникающее при тангенциальных движениях поверхности, оказывает резко тормозящее действие на эти движения. К таким веществам относятся агар-агар, желатин, столярный клей и др. Кроме того, максимумы первого рода устраняются применением достаточно концентрированных фонов.
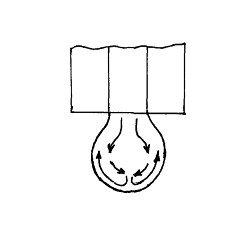
Рис. 3. Схема движения ртути в ртутной капле.
Наличие в растворе кислорода искажает полярограммы, так как он восстанавливается на катоде и дает две полярографические волны.
Одна волна (от 0,15 до 0,2 В) соответствует восстановлению кислорода до пероксида водорода, а другая (от - 1,7 до - 1,3 В) - восстановлению пероксида водорода до воды или группы ОН-. Кислород удаляют из испытуемого раствора пропусканием через него в течение 10...20 минут инертного газа (Ar, N2, He) или введением в раствор со щелочной или нейтральной средой сульфита натрия до 1г. Восстановление О2 и окисление сульфита до сульфата происходит в течение 2...5 минут.
Полярография с РКЭ характеризуется рядом достоинств:
1) чистота электрода вследствие постоянного обновления электрода в процессе капания ртути;
2) процессу анализа на РКЭ, по сравнению с платиновым электродом, практически не мешает восстановление ионов водорода;
3) возможность анализа растворов с малой концентрацией исследуемых веществ приблизительно (1...5) 10-5 моль/л и из пробы объемом до 1 мл;
4) классической полярографией можно обнаружить менее 0,01 мг исследуемого вещества с погрешностью 2%.
Основными недостатками классической полярографии являются:
1) невозможность использования для анализа веществ, которые подвергаются только окислению, а не восстановлению, т.е. при потенциалах положительнее равновесного потенциала ртути в данном растворе ( 0,2 В);
2) невозможность резкого увеличения чувствительности из-за искажающего влияния тока заряжения РКЭ;
3) токсичность и неудобство в работе (например, в полевых условиях);
4) относительно большое время измерения (от 3 до 10 мин для одной пробы раствора).
Налаживание производств новых видов сверхчистых материалов, а также возрастающая острота экологических проблем потребовали разработки более чувствительных методов анализа, позволяющих обнаружить различные примеси с концентрацией 10-8 моль/л.
Для уменьшения влияния тока заряжения в современных полярографах предусмотрены электрические схемы для автоматической его компенсации и для непосредственной записи фарадеевского тока. Однако точность такой компенсации ограничена, особенно при малых концентрациях реагирующего вещества.
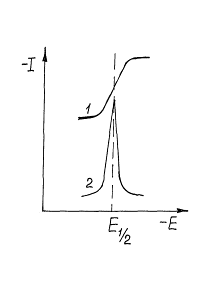
Рис. 4. Интегральная (1) и дифференциальная (2) полярограммы.
В дифференциальной полярографии чувствительность и селективность классической полярографии можно повысить, если регистрировать зависимость от потенциала не тока, а его производной от потенциала (dI/dE). В этом случае вместо полярографической волны получают кривую с максимумом (рис.2.14.3). Потенциал максимума соответствует потенциалу полуволны обычной полярограммы, а высота максимума пропорциональна концентрации исследуемого вещества. Сигнал, пропорциональный производной тока от потенциала, формируется в полярографе с помощью сравнительно простой электрической схемы. Для увеличения отношения полного сигнала (фарадеевский ток) к фону (ток заряжения) можно воспользоваться разной временной зависимостью фарадеевского тока Iф и тока заряжения Iз при росте ртутной капли. Установлено, что Iф растет во времени пропорционально t1/6, а ток заряжения падает пропорционально t-1/3. Таким образом, при отрыве капли соотношение Iф к Iз максимально и условия измерения наиболее благоприятны. В методе так называемой таст-полярографии (от нем. tasten - зондировать) измерения тока проводят не непосредственно во время “ жизни “ капли, а только в течение короткого времени - 5...20 мс - перед отрывом капли. Этим способом удается чувствительность метода увеличить на порядок, т.е. до (1...5) 10-6 моль/л.
Амперометрическое титрование - это метод анализа, возникший на основе классической полярографии. В нем предельный диффузионный ток используют для нахождения точки эквивалентности при проведении титрования. Амперометрическое титрование возможно только при условии электроактивности (способности разряжаться на электроде) определяемого вещества или вещества титранта или хотя бы одного продукта их реакции. Для проведения амперометрического титрования аликвотную часть анализируемого раствора помещают в электролизер. В раствор опускают РКЭ или твердый (чаще всего платиновый) микроэлектрод и электрод сравнения. На электроды подают напряжение, отвечающее области предельного тока электроактивного вещества, и проводят титрование. После добавления из бюретки каждой отдельной порции титранта отмечают силу тока.
Пусть электроактивным является определяемое вещество, например, Pb2+, тогда добавление титранта (H2SO4), реагирующего с ним, будет уменьшать концентрацию определяемого вещества в растворе, в соответствии с этим будет уменьшаться и предельный диффузионный ток.
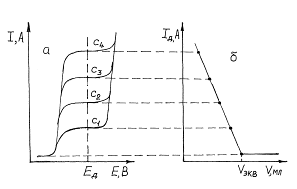
Рис. 5. Схема получения кривой амперометрического титрования (б) электроактивного определяемого вещества по его полярограммам (а) при концентрациях ![]() .
.
Поскольку предельный диффузионный ток, согласно уравнению Iд = КС, пропорционален концентрации, то амперометрические кривые титрования, построенные в координатах Iд - V, являются линейными (рис.2.14.4). По ним графически находят объем титранта в точке эквивалентности (Vэкв).
Значение напряжения, при котором следует проводить титрование, предварительно устанавливают по полярограммам анализируемого раствора, титранта или раствора продуктов реакции.
Если электроактивным является вещество титранта, то ток останется практически равным нулю, пока не будет достигнута конечная точка титрования и не появится в растворе избыток титранта. Начиная с этого момента, ток будет расти. Такую кривую титрования можно получить, напри - мер, при титровании ионов цинка раствором K4 [Fe(CN) 6], окисляющегося на платиновом электроде (рис. 6, а).
Титрование возможно, если электроактивен образующийся продукт реакции определяемого вещества и титранта. Кривая титрования тогда имеет вид, изображенный на рис. 6, б.
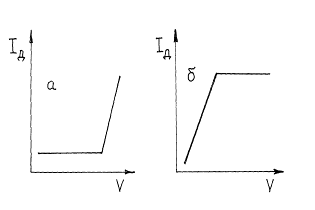
Рис. 6. Кривые амперометрического титрования при электроактивном титранте (а) или продукта реакции титрования (б).
Для амперометрического титрования пригодны реакции комплексообразования, осаждения, а также редоксиреакции. Концентрация титруемого раствора обычно порядка 10-3 моль/л. Определять можно не только неорганические, но и органические вещества (например, фенолы). Метод значительно проще, экспресснее и доступнее полярографии и вольтам перометрии.
Инверсионная вольтамперометрия (вольтамперометрия с накоплением) - это метод, отличающийся от других не формой используемого импульса, а принципом проведения анализа. Исследуемое вещество сначала частично или полностью осаждают электрохимическим путем из пробы раствора на инертный электрод-подложку (стадия накопления, концентрирования). Чаще всего этот метод применяют для катионов металлов, которые катодно осаждают на стационарном (не капающем!) ртутном электроде или на платиновых, золотых, графитовых и т.д. твердых индифферентных электродах. На второй стадии, после извлечения ионов из раствора, электрод с пленкой осажденного металла подвергают анодной поляризации с линейно изменяющимся (с постоянной скоростью) потенциалом.
При превышении в ходе поляризации равновесного значения потенциала, отвечающего определяемому металлу, он начинает растворяться, причем тем активнее, чем больше поляризующее напряжение. Соответственно с этим растет регистрируемый самописцем-потенциометром ток растворения. Выходная кривая (вольтамперограмма) данного метода имеет вид кривой с максимумом, отвечающим окончанию растворения концентрата и выходу фронта растворения на поверхность индифферентного электрода (рис.2.14.6).
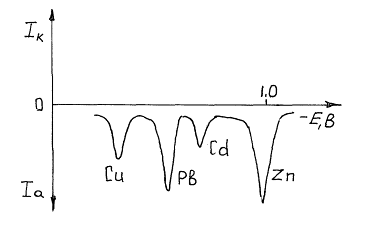
Рис. 6, б. Инверсионная вольтамперограмма раствора, содержащего цинк, кадмий, свинец и медь (фон 0,1 М НСООН, Енакоп =1,2 В).
Положение и высота максимумов тока на вольтамперограмме характеризуют как природу, так и общее количество (концентрацию) исследуемого вещества. В присутствии нескольких определяемых веществ в специально подобранных условиях (фоновый электролит, потенциал накопления и т.д.) на кривой можно получить несколько максимумов тока, отвечающих каждому веществу. Данный метод чрезвычайно чувствителен - в отдельных случаях могут быть обнаружены примеси металлов с концентрацией 10-9 моль/л и даже ниже. Необходимо иметь в виду, что при очень малых концентрациях для полного осаждения ионов из раствора требуется значительное время накопления - иногда до одного часа, но этот недостаток нивелируется простотой применяемого оборудования. Для определения неизвестной концентрации применяют в основном метод стандартной серии или метод добавок.
Электрогравиметрия (ЭГМ) является разновидностью гравиметрии. Особенность ЭГМ заключается в осаждении определяемого элемента путем электролиза на предварительно взвешенном электроде. О массе элемента в растворе судят по увеличению массы электрода после электролиза.
ЭГМ применяют для определения металлов из растворов, в которых они присутствуют в виде ионов.
При электролизе катионы перемещаются к катоду, выделяясь на нем в виде металлов. Только очень немногие металлы осаждаются на аноде. К ним относятся, например, Mn и Pb, окисляющиеся в процессе электролиза до MnO2 и PbO2.
ЭГМ применяют для определения металлов, дающих плотные осадки на электроде, не осыпающиеся при промывании, высушивании и взвешивании. Кроме того, ЭГМ применяют только в тех случаях, когда осаждение определяемого металла не сопровождается соосаждением других металлов или примесей.
Электроды, применяемые в ЭГМ, должны отвечать следующим требованиям:
1) быть химически инертными;
2) хорошо удерживать образующиеся осадки;
3) иметь возможно меньшую массу и возможно большую поверхность;
4) не препятствовать перемешиванию раствора.
Всем этим требованиям в наибольшей степени удовлетворяют платиновые сетчатые электроды. Анодом, в большинстве случаев, служит платиновая проволока, согнутая в спираль.
Для проведения ЭГМ два платиновых электрода погружают в стакан с анализируемым раствором, подсоединяют электроды к внешнему источнику тока и проводят электролиз. При прохождении тока через раствор электролита происходят процессы восстановления и окисления соответствующих веществ на электродах. Связь между количествами веществ, участвующих в электродных процессах, и количеством электричества Q (Q = It) через цепь за время электролиза t при токе I устанавливается двумя законами Фарадея:
1) масса вещества, выделившаяся при электролизе, пропорциональна количеству электричества, прошедшего через раствор;
2) при прохождении через раствор одного и того же количества электричества на электродах выделяется одно и то же количество вещества эквивалента.
Математически оба закона можно представить формулой
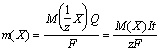 ,
,
где m(X) - масса вещества X, выделившегося при электролизе;
M(1/z X) и M(X) - молярная масса эквивалента и молярная масса вещества X, соответственно;
z - число эквивалентности;
F - число Фарадея, равное количеству электричества (96500 Кл), которое требуется для выделения 1 моль эквивалентов вещества.
Формула позволяет решать различные задачи, связанные с электролизом. Например, вычислить продолжительность при заданной силе тока для выделения определенной массы вещества. На практике электролиз требует больше времени, чем это следует из формулы. Это связано с побочными реакциями, обычно сопровождающими главные. Поэтому КПД тока, иначе называемый выходом по току, почти всегда ниже 100%.
Выход по току может быть определен как отношения массы вещества m, реально полученного при электролизе, к массе вещества, которая могла бы получиться в соответствии с законом Фарадея m0, если бы количество электричества не расходовалось на побочные процессы:
![]() .
.
При прохождении через раствор электрического тока на электродах выделяются продукты электролиза, что приводит к возникновению в системе ЭДС обратной внешней ЭДС источника тока. Это явление называется электрохимической поляризацией, а возникающая обратная ЭДС - ЭДС поляризации. Ее можно заметно уменьшить, прибавляя так называемые деполяризаторы, т.е. вещества, разрежающиеся прежде, чем ионы, которые разрежались бы в их отсутствие.
Таким образом, чтобы электролиз мог происходить, необходимо приложить к электродам напряжение, превышающее ЭДС поляризации. Наименьшее напряжение, которое необходимо приложить к электродам для того, чтобы вызвать непрерывный электролиз данного электролита, называется его напряжением разложения Ер. Ер должно быть больше ЭДС гальванического элемента Е (Е = Еа - Ек) на величину перенапряжения Ер = Е + = (Еа+ a) - (Ек - k), где Еа и Ек - равновесные потенциалы анода и катода, а а и к - перенапряжения на аноде и катоде.
Величина перенапряжения зависит от:
1) плотности тока j = I/S (где S - площадь поверхности электрода). Чем больше j, тем больше ;
2) состояния поверхности электрода: на гладком электроде больше, чем на шершавом, так как при одинаковой силе тока приходящаяся на единицу поверхности плотность тока больше;
3) температуры: повышение температуры уменьшает ;
4) природы электрода и различных примесей в растворе.
При электролизе нужно учитывать силу тока в цепи. Чем больше I, тем больше j и тем больше в единицу времени на поверхности электрода выделится определяемого металла. Следовательно, тем быстрее закончится электролиз и анализ в целом.
Однако при слишком большой j осадок получается рыхлым (губчатым), непрочно связанным с электродом. Причина этого в том, что при слишком большой j скорость разрядки ионов определяемого металла становится больше скорости их подвода к электроду. Поэтому раствор около катода начинает настолько обедняться ионами, что на катоде начинает восстанавливаться водород, пузырьки которого разрыхляют осадок. Введение комплексообразующих компонентов предотвращает выделение водорода и способствует получению прочных однородных осадков металлов.
Многие металлы, например Zn, Sn, Pb, при низких плотностях тока выделяются в виде непрочного слоя. Предполагается, что причина этого - присутствие в электролите растворенного кислорода и примесей окислителя.
Условия электролиза должны быть выбраны так, чтобы происходило выделение только одного металла, а не их смеси, и чтобы выход по току составлял 100%.
После электролиза электроды промывают несколько раз дистиллированной водой, не отключая электроды от источника тока, затем сушат и точно взвешивают. По разности масс электродов, без осадка и с ним, находят массу определяемого вещества в растворе.
Внутренний электролиз ЭГМ можно выполнить в накоротко замкнутом гальваническом элементе. При этом не требуется внешнего источника тока, так как осадок выделяется за счет энергии гальванического элемента. Такой вариант ЭГМ называют внутренним электролизом.
Библиографический список
1. Янсон Э.О. Теоретические основы аналитической химии: Учеб. для хим. фак. ун-тов.2-е изд., перераб. и доп. - М.: Высш. шк., 1987.
2. Основы аналитической химии: Учеб. для вузов / Под ред. Ю.А. Золотова. В 2кн. Кн.1. Общие вопросы. Методы разделения. Кн.2. Методы химического анализа. - М.: Высш. шк., 1999.
3. Аналитическая химия. Химические методы анализа / Под ред.О.М. Петрухина. - М.: Химия, 1992.
4. Bacильeв B.П. Aнaлитичecкaя xимия: Учeб. для xим.-тexнoл. cпeц. вyзoв: B 2 ч. - M.: Bыcш. шк., 1989.
5. Пилипeнкo A. T., Пятницкий И. B. Aнaлитичecкaя xимия: Учeб. для cтyд. xим. cпeц. yн-тoв: B 2 кн. - M.: Xимия, 1990.
6. Пocыпaйкo B.И. и дp. Xимичecкиe мeтoды aнaлизa: Учeб. пoc. для xим. -тexнол. вyзoв. - M.: Bыcш. шк., 1989.
7. Aлeкceeв B. H. Koличecтвeнный aнaлиз. - M.: Xимия, 1972.
8. Дорохова Е. Н, Прохорова Г.В. Аналитическая химия. Физико-химические методы анализа: Учеб. для почвенно-агрохим. спец. ун-тов и вузов. - М.: Высш. шк., 1991.
9. Толстоусов В.Н., Эфрос С.М. Задачник по количественному анализу. Л.: Химия, 1986.
10. Чapыкoв A. K. Maтeмaтичecкaя oбpaбoткa peзyльтaтoв xимичecкoгo aнaлизa. П.: Xимия, 1984.
11. Справочник химика-аналитика. М.: Металлургия, 1976.
12. Лурье Ю.Ю. Справочник по аналитической химии. Изд.5-е, перераб. и доп. М.: Химия, 1979.
0 комментариев