Связь автоускорения при радикальной полимеризации метилметакрилата в массе со структурными изменениями полимеризующейся системы
Известно, что радикальная полимеризация в массе многих мономеров, в том числе такого важного в промышленном отношении мономера, как метилметакрилат (ММА), сопровождается автоускорением (гель-эффект). Этот эффект проявляется в одновременном резком увеличении скорости реакции и молекулярных масс образующегося ПММА при достижении некоторой степени превращения (т. е. концентрации полимера в реакционной системе). Несмотря на растущее год от года число работ, посвященных гель- эффекту, в литературе пока еще нет единой точки зрения о природе этого явления [1 — 4]. Хорошо известно, что реологические методы весьма информативны при изучении свойств концентрированных растворов полимеров. По этой причине в последние годы появился ряд работ, в которых исследуются реологические характеристики полимеризующихся систем [5, 6]. В настоящей работе непосредственно в ходе радикальной полимеризации ММА в массе измерены реологические параметры (наибольшая ньютоновская вязкость "По и начальный модуль высокоэластичности G0) реакционной системы с целью установления корреляции кинетических параметров начала автоускорения со структурой и физическим состоянием полимеризующейся системы, представляющей собой растворы ПММА различных концентраций и ММ в собственном мономере.
Технический ММА очищали по общепринятой методике. Полимеризацию инициировали разложением перекиси лаурила (ПЛ) при 60°; в качестве регулятора ММ образующегося ПММА кроме инициатора использовали также лаурилмеркаптан (ЛМ). Кинетические измерения проводили на микрокалориметре ДАК-1-1 в изотермических условиях.
Реологические измерения осуществлялись на ротационном -вискозиметре постоянных напряжений сдвига ВНП-01 (типа описанного в работе [7]), позволяющем охватить широкие диапазоны напряжений (от 4-Ю-1 до 4-103 Па), скоростей сдвига (от 4-102 до Ю-5 с-1) и проводить измерения вязкостей в интервале от 10~3 до 107 Па/с. Прибор позволял производить измерения в рабочем узле с гидрозатвором в инертной атмосфере аргона. Все реологические параметры получены при низких напряжениях сдвига (0,4 — 0,7 Па), что обеспечивало измерения наибольшей ньютоновской вязкости и начального модуля высокоэластичности, рассчитываемого из данных по «упругому отскоку» образующихся в ходе полимеризации растворов полимера в мономере.
Реакционные системы подбирали таким образом, чтобы за время измерения не происходило существенных изменений в состоянии системы. Так, в области максимальной скорости полимеризации изменения состава системы за время измерения не превышали 0,3%, что позволило определять вязкости с ошибкой не более 10—15%. Реакционные смеси ММА с ПЛ и ЛМ для кинетических и реологических измерений освобождали от растворенного кислорода воздуха барботированием аргона в течение 30 мин. Все кинетические и реологические измерения проводили при 60°. Для удобства сопоставления результатов кинетических и реологических измерений концентрацию полимера с во всех случаях выражали в вес, %, таким образом, степень превращения полимеризации или процент конверсии q совпадал с величиной с. Концентрационные зависимости вязкости полимеризующихся систем разбивали на отдельные прямолинейные участки при помощи статистической обработки экспериментальных данных по методу наименьших квадратов.
Молекулярные массы ПММА рассчитывали из соотношения [т]]=8,69-10~5 Д?°.78 18] (бензол, 30°).
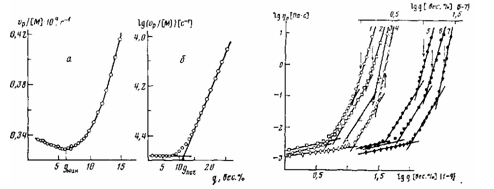
Тис. 1. Зависимость приведенной скорости полимеризации (о) и ее логарифма (б) от глубины превращения ММА. [ГШ] =5-10-3 моль/л; 60°
Рис. 2. Зависимость логарифма вязкости реакционных систем от глубины превращения ММА при 60^ 1 — Af„=324 ООО; 2 - [ЛМ]=3-10-3 моль/л, J0W98OOO; 3-ГЛМ]=10^ моль/л, Af„=18 000; 4-модельная система ММА+ПММА, ЛГ„=79 400, 50°, [14]; 5- Af„=495ООО; 6- [ЛМ]=3,3-10-3 моль/л, Ж„=170ООО; 7- [ЛМ]=10 моль/л, JW„=97 000. [ПЛ] = 10-2 (1-3) и 5-Ю-3 моль/л (5-7). Значения Mv приведены для ПММА, образующегося в системе до конверсии
Начало автоускорения радикальной полимеризации определяют различными способами. Некоторые авторы отождествляют его с конверсией, при которой происходит заметное (~2 — 3%) отклонение от прямолинейной зависимости степени превращения от времени реакции. Большинство же исследователей, основываясь на том, что скорость полимеризации имеет первый порядок по концентрации мономера [9], определяют конверсию наступления автоускорения qnor по перелому зависимости логарифма приведенной скорости полимеризации lgvp/[M], где vp — скорость реакции, а [М] — текущая концентрация мономера, от конверсии (рис. 1,6). Следует отметить, что эти два метода определения дают близкие значения конверсии, соответствующей началу автоускорения. Поэтому в дальнейшем мы будем пользоваться величиной длог. Недавно авторами работы [10] было предложено считать началом автоускорения конверсию минимума приведенной скорости полимеризации (рис. 1, о), существование которого предположительно объяснялось равенством вкладов сегментальной и трансляционной подвижности макромолекул в константу скорости бимолекулярного обрыва цепи. Из полученных в настоящей работе кинетических данных полимеризации ММА в присутствии различных концентраций инициатора и передатчика цепи, изменяющих величину ММ образующегося полимера, следует, что во всех случаях длог больше дМин, причем увеличение ММ образующегося в ходе реакции ПММА сопровождается уменьшением значений обеих характеристических конверсии. Ранее [4] нами для случая полимеризации ММА в массе, была установлена следующая зависимость между степенью превращения начала автоускорения и средневязкостной степенью полимеризации образующегося к этой конверсии полимера
![]()
На рис. 2 приведены зависимости логарифма вязкости реакционных систем от степени превращения полимеризующегося в массе ММА в условиях образования полимера с различными ММ. Видно, что темп увеличения
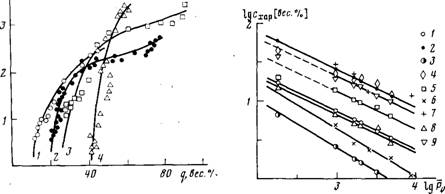
Рис. 3. Зависимость логарифма начального модуля высокоэластичности реакционных систем от глубины превращения ММА при 60°: 1 - М„=495 ООО; 2 - MV=32A ООО; 3 — [ЛМ]=3-10-3 моль/л, Af„=198000; 4-[ЛМ] = 10"1 моль/л, Vtf„=18000.
(1) и 10-2 моль/л (2 - 4)
Рис. 4. Зависимость характеристических концентраций ПММА в ММА в полимери-зующихся и модельных системах от степени полимеризации полимера, образующегося к началу автоускорения. Значения с определяли из соотношения (2) со значениями параметра А, соответствующими моделям [23] и [27] (1, 2), из величин 1/|п| (3), по значениям длог и дмин соответственно (4, 5), методом светорассеяния по данным работы [19] (6, 7) и по первому и второму излому на концентрационных зависимостях вязкости рис. 2 (8, 9)
вязкости возрастает с увеличением конверсии в пределах одной кривой и с увеличением ММ образующегося полимера для различных кривых. На этом же рисунке для сравнения приведены значения вязкостей растворов ПММА в ММА, приготовленных смешением компонентов, по данным [14]. Из сопоставления этих данных следует, что в изученном интервале концентраций полимера возрастание вязкости в системе в ходе полимеризации аналогично изменению вязкости модельных растворов «мертвого» полимера соответствующей ММ в собственном мономере. Несколько неожиданным явилось существование двух изломов, наблюдаемых на кривых зависимости вязкости от концентрации полимера в логарифмических координатах. Ряд авторов при исследовании реологических свойств растворов полимеров [15, 16] представляет логарифмические зависимости вязкости от концентрации полимера в системе в виде двух прямых с тангенсами угла наклона, равными ~1 и ~5, пересекающихся при некоторой критической концентрации. Для жесткоцепных макромолекул [17] отмечается, что в области высоких концентраций полимера тангенс угла наклона постепенно возрастает до 30—40. Применяемые авторами методики измерения вязкости и приготовления растворов из-за ограниченного в силу трудоемкости количества концентрационных точек не позволяли детально проанализировать изменение вязкости в зависимости от состава раствора. Методика, используемая в нашей работе, предоставила возможность пройти практически непрерывный ряд концентраций>полимера в растворе, а следовательно, и значений вязкости реакционной системы в ходе полимеризации. Это позволило разбить кривую зависимости lg r|— lg g в изученном интервале степеней превращения (д<60%) на три прямолинейных участка (рис. 2). Соответствующие прямые имеют различные углы наклона, тангенсы которых равны на начальном участке 0,7±0,1, на среднем участке 3,5±0,5 и на конечном участке >7. При этом на концентрационных зависимостях вязкости были выделены две характеристические степени превращения — qt и q2, соответствующие первому и второму излому. Существование участков зависимости lg r—lg q с тангенсами угла наклона, равными 1 и 3,5, следует из представлений, выдвинутых в работе [18].
На рис. 3 приведены зависимости lg G0 от q, которые представляют собой плавные кривые с характерным резким уменьшением модуля высокоэластичности при снижении концентрации полимера в системе. Минимальные степени превращения, при которых удавалось измерить начальный модуль высокоэластичности (т. е. соответствующие началу проявления реакционной системой высокоэластичности) указаны стрелками на концентрационных зависимостях вязкости (рис. 2). Важно, что их положение практически совпадает со вторым изломом на этих кривых.
Представляло интерес выяснить, будут ли характеристические концентрации полимера в системе, фиксируемые при измерении реологических параметров полимеризатов, подчиняться зависимости типа (1). Положительный ответ на этот вопрос дает рис. 4, на котором в логарифмических координатах представлены результаты определения кинетическим и реологическим методами характеристических конверсии, полученные в данной работе. Для обеих характеристических концентраций полимера в реакционной системе, фиксируемых реологически, параметр а уравнения (1) равен 0,5. Постоянная К для первого перехода равна 215, для второго — 675. Кроме того, на рис. 4 приведены результаты определения характеристических концентраций полимера в растворе, полученные нами ранее [19, 20] другими методами, а также характеристические концентрации, рассчитанные теоретически. Видно, что все зависимости характеристических концентраций от степени полимеризации образующегося полимера, построенные в логарифмических координатах, укладываются на прямые, незначительно отличающиеся углами наклона (следовательно, имеющие близкие значения параметра а) и смещенные относительно друг друга по шкале концентраций (т. е. различающиеся величиной отрезка, отсекаемого на оси ординат, определяющего значение К). Все прямые рис. 4 можно разделить по углу наклона и их относительному расположению по шкале концентраций на три группы, которые и будут охарактеризованы ниже.
Рассмотрим в этой связи последовательность структурных превращений в полимеризующейся системе в ходе реакции (т. е. с увеличением концентрации полимера) и проанализируем возможную связь этих превращений с кинетикой полимеризации. Известно, что первой естественной границей при увеличении концентрации полимера является переход от разбавленного к умеренно концентрированному раствору при критической концентрации перекрывания макромолекулярных клубков. Эта концентрация определяется средней концентрацией звеньев макромолекулы в объеме изолированного полимерного клубка
![]() (2)
(2)
где_Р„ — среднечисленная степень полимеризации растворенного полимера, a R — радиус инерции. В свою очередь
![]() (3)
(3)
где v — степенной показатель исключенного объема, равный 0,6 для хорошего растворителя и 0,5 для б-растворителя. Из выражений (2) и (3) следует, что
![]() (4)
(4)
т.е. получается уравнение типа (1), в котором степенной показатель при Рп изменяется в пределах от 0,5 до 0,8 в зависимости от термодинамического качества растворителя, а величина const определяется множителями в соотношениях (2) и (3).
Простейшим экспериментальным способом определения с* является измерение характеристической вязкости данного полимера в данном растворителе. Измерения характеристических вязкостей ПММА в ММА при 60° были проведены нами ранее [19], а соответствующие результаты (в простейшем предположении, что с*=1/[п]) представлены нижней прямой на рис. 4. В работе [19] мы наблюдали переход от разбавленного к умеренно концентрированному раствору в ходе полимеризации ММА также и методом светорассеяния. Было показано, что интенсивность рассеяния реакционными системами под углом 90° проходит через максимум при степенях превращения, соответствующих с*. При изменении ММ образующегося ПММА положение максимума интенсивности смещается по шкале конверсии, а соответствующие им точки также ложатся на прямую в координатах рис. 4, расположенную несколько выше прямой 1/[п]. Характерно, что углы наклона этих двух прямых, составляющих первую группу, отличаются от углов наклона других прямых рис. 4 и равны между собой. Их тангенс наклона составляет 0,6 (т. е. 3v - 1=0,6). Последнее обстоятельство свидетельствует о том, что ММА как растворитель для ПММА уже в разбавленном растворе приближается к растворителю. Следует отметить, что ни кинетика полимеризации, ни реологические измерения в системе не чувствительны к переходу от разбавленного к умеренно концентрированному раствору, по-видимому, вследствие того, что при этих концентрациях еще не происходит изменения характера диффузии полимерных клубков.
Последующее увеличение концентрации полимера в реакционной смеси выше некоторого другого характеристического значения сопровождается увеличением темпа нарастания вязкости (т. е. переломом на реологической кривой) и появлением минимума скорости полимеризации на дифференциальной кинетической кривой. Обе характеристические концентрации, фиксируемые кинетическим и реологическим методами, подчиняются уравнению типа (1) и составляют вторую группу прямых на рис. 4.
Известно, что при концентрациях полимера в растворе выше концентрации заполнения объема набухшими клубками происходит уменьшение среднего размера клубков до невозмущенного 9-размера и их частичное взаимное проникновение [21]. Характеристические концентрации полимера в растворе, при которых заполняется весь объем эквивалентными клубками, поджатыми до 8-размера, и выравниваются плотности сегментов в растворе, могут быть вычислены из уравнения (5), полученного рядом авторов и проанализированного в работе [18]
![]()
где NA — число Авогадро, Re — средний радиус инерции невозмущенного клубка, параметр А, изменяющийся в пределах ~0,2—1,0, зависит от выбора радиуса эквивалентной сферы полимерных клубков в растворе и от характера их упаковки.
Уравнение (5) фактически эквивалентно соотношению (2). Представляло интерес сопоставить концентрации полимера в реакционной системе, при которых происходят переходы, фиксируемые реологическим и кинетическим методами, с характеристическими концентрациями, предсказываемыми уравнением (5). Последние были рассчитаны при использовании усредненного значения (Re/M4') =580- 10~и см [22] для ПММА и предположения о плотнейшей гексагональной упаковке эквивалентных Э-сфер (когда А =0,18 [23]). Расчетные значения практически совпадают с концентрациями, соответствующими первому перелому на реологических кривых, и примерно вдвое меньше концентраций, соответствующих минимумам приведенной скорости полимеризации (рис. 4). Соприкосновение невозмущенных клубков при их концентрировании приводит к повышению темпа роста вязкости реакционных систем при увеличении концентрации. Этот эффект, по-видимому, проявляется и в кинетике полимеризации в виде минимума скорости реакции, но при несколько больших концентрациях полимера. В работе [13] было высказано предположение о том, что минимум скорости реакции наблюдается при конверсиях, еще недостаточных для обнаружения отклонения от линейной зависимости вязкости от степени превращения. Проведенный нами эксперимент для системы ММА — ПММА доказывает обратное. Дальнейшее увеличение степени превращения должно приводить к взаимному проникновению сжатых до О-размера клубков ПММА, к их перепутыванию с образованием устойчивой и однородной по объему сетки зацеплений. Этот переход фиксируется одновременно как реологически (по дополнительному увеличению темпа нарастания вязкости и обнаружению высокоэластической деформации полимеризующейся системы), так и кинетически (по резкому увеличению скорости полимеризации). На кривых зависимостей светорассеяния от степени превращения в этой области концентраций ранее наблюдали выход на участок линейного уменьшения интенсивности рассеяния [19]. Соответствующие значения характеристических конверсии, полученные различными методами, в логарифмических координатах (рис. 4) ложатся на близко расположенные прямые с тангенсом угла наклона, равным 0,5 (третья группа прямых). Следует отметить, что уравнение (5) позволяет количественно описать характеристические концентрации дЛ0Г и концентрации, соответствующие перелому на реологических зависимостях, как функции степени полимеризации образующегося полимера, если принять, что радиус эквивалентной сферы полимерного клубка ДЭКв=0,665/?в (т. е. параметр А в уравнении (5) равен 0,6). Указанное соотношение между RBKB и Re было получено в теории Кирквуда — Райзмана при расчете коэффициента трения макромолекул [24]. Позднее оно было использовано в работах Оноги для расчета характеристических концентраций при исследовании реологических свойств концентрированных растворов полимеров [25] и в работах Тернера для определения конверсии наступления автоускорения [3]. Можно полагать, что изменение кинетической и реологической зависимостей, наблюдаемые при формировании сетки зацеплений, связаны с переходом от обычного к «рептационному» характеру диффузии макромолекул [26, 27]. Действительно, модель, связывающая наступление гель-эффекта с переходом от трехмерной диффузии макромолекул к одномерной («рептация»), позволила оценить константу К в уравнении (1), описывающем наступление автоускорения при радикальной полимеризации ММА. Эта величина оказалась порядка 103 в предположении α=0,5 [28]. Последующее увеличение степени превращения обусловливает дальнейшее снижение скорости бимолекулярного обрыва за счет увеличения плотности сетки зацеплений и не должно приводить к качественным изменениям характера диффузии реагирующих частиц в реакционной системе вплоть до наступления стеклования, когда и реакция роста цепи переходит в диффузионно-контролируемый режим. Следует отметить, что ни один из переходов на кинетических и реологических кривых не может быть охарактеризован универсальным значением вязкости или свободного объема реакционных систем.
Таким образом, результаты, полученные независимыми физико-химическими методами (кинетическим, гидродинамическим, оптическим), позволяют считать, что автоускорение радикальной полимеризации ММА связано с комплексом структурно-физических переходов полимеризационной системы, обусловливающих изменение характера диффузии растущих полимерных цепей.
Литература
1. Gardenas J.N.. O'Driscoll К.F.J. Polymer Sci. Polymer Chem. Ed., 1976, v. 14, № 4, p. 883.
2. Marten F.L., Hamielec A.E. Amer. Chem. Soc. Symp. Ser., 1979, № 104, p. 43.
3. Turner D.T. Macromolecules, 1977, v. 10, № 2, p. 211.
4. Lachinov M.В., Simonian R.A., Georgieva T.D., Zubov V.P., Kabanov V.A.J. Polymer Sci. Polymer Chem. Ed., 1979, v. 17, № 2, p. 613.
5. Емельянов Д.H., Кононова И.Е., Рябов А.В., Симонова Н.Ф. Высокомолек. соед. Б, 1975, т. 17, № 2, с. 163.
6. Куличихин С.Г., Малкин А.Я. Высокомолек. соед. А, 1980, т. 22, № 9, с. 2093.
7. Васильев В.Г., Козлов О.А., Константинов А.А., Крашенинников С.К., Роговина Л.3., Слонимский Г.Л. Коллоидн. ж., 1977, т 39, № 5, с. 938.
8. Fox Т.G., Kisinger J.В., Mason H.F., Shuele E.M. Polymer, 1962, v. 3, № 1, p. 72.
9. Schulz G.V., Harborth G. Maeromolec. Chem., 1947, v. 1, p. 106.
10. Dionosio J., Mahabadi H.K., O'Driscoll K.F., Abuin E., Lissi E.A.J. Polymer Sci. Polymer Chem. Ed., 1979, v. 17, № 7, p. 1891.
11. Lee M.В., Turner D.T. Polymer Preprints, 1977, v. 18, № 2, p. 539.
12. Lee H.В., Turner D.T. Polymer Preprints 1978, v. 19, № 2, p. 603.
13. Dionisio J.M., O'Driscoll K.F.J. Polymer Sci. Polymer Chem. Ed., 1980, v. 18, № 11, p. 3199.
14. Brooks B.W. Proc. Roy. Soc, 1977, v. A357, № 1, p. 183.
15. Porter R.S., Johnson J.F. Chem. Rev., 1966, v. 66, № 1, p. 1.
16. Дрееаль В.E. Дис. на соискание уч. ст. докт. хим. наук. М.: НИФХИ им. Л.Я. Карпова, 1974, 304 с.
17. Виноградов Г.В., Малкин А.Я. Реология полимеров. М.: Химия, 1977, с. 209.
18. Berry G.С, Nakayasu Н., Fox Т.G.J. Polymer Sci. Polymer. Phys. Ed., 1979, v. 17, № 11, p. 1825.
19. Симонян P.А., Касаикин В.А., Лачинов M.В., Зубов В.П., Кабанов В.А. Докл. АН СССР, 1974, т. 213, № 3, с. 631.
20. Лачинов М.В., Дрееаль В.Е., Касаикин В.А., Симонян Р.А., Шипулина Н.И., Зубов В.П., Кабанов В.А. Высокомолек. соед. А, 1977, т. 19, № 4, с. 741.
21. Daond М., Cotton J.P., Farnoux В., Jannink G., Sarma G., Benoit H., Duplessi R., Picot C, de Gennes P. G. Macromolecules, 1975, v. 8, № 6, p. 804.
22. Polymer Handbook/Ed. Brandrup J., Immergut E.N.N.Y.: Interscience, 1975.
23. Berry G.C.J. Phys. Chem., 1966, v. 70, № 4, p. 1194.
24. Klrwood J.G., Riseman J.J. Chem. Phys., 1948, v. 16, № 6, p. 565.
25. Onogi S., Masuda Т., Miyanaga N., Kimura J.J. Polymer Sci. A, 1967, v. 5, № 5, p. 899.
26. de Gennes P.G. Macromolecules, 1976, v. 9, № 4, p. 587.
27. Klein J. Macromolecules, 1978, v. 11, № 5, p. 852.
28. Битюрин H.M., Генкин В.И., Зубов В.П., Лачинов М.В. Высокомолек. соед. А, 1981, т. 23, № 8, с. 1702.
0 комментариев