Министерство образования и науки Российской Федерации
Федеральное государственное бюджетное образовательное
учреждение высшего профессионального образования
«Петрозаводский Государственный Университет»
(ПетрГУ)
Эколого-биологический факультет
Кафедра молекулярной биологии, биологической и органической химии
Прокопенко
Христина Николаевна
Специальность
«Биология»
АКТИВНОСТЬ КАЛЬЦИЙЗАВИСИМЫХ ПРОТЕИНАЗ В МОЗГЕ КРЫС
С ЭКСПЕРИМЕНТАЛЬНОЙ НЕЙРОПАТОЛОГИЕЙ
Дипломная работа
Научный руководитель:
кандидат биологических наук,
ведущий научный сотрудник ИБ КарНЦ РАН
Лысенко Людмила Александровна
Петрозаводск, 2015.
Содержание
ВВЕДЕНИЕ
ГЛАВА 1.ОБЗОР ЛИТЕРАТУРЫ
1.1 Кальпаин/кальпастатиновая протеолитическая система
1.2 Биохимические механизмы развития нейродегенеративных заболеваний
ГЛАВА 2.МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
2.1 Реагенты и приборы
2.2 Моделирование нейродегенерации у лабораторных животных
2.3 Анализ биохимических показателей
2.3.1 Экстракция белков из тканей
2.3.2 Определение активности кальпаинов
2.3.3 Зимография с казеином
2.3.4 Электрофорез белков полиакриламидном геле
2.3.5 Вестерн-блот анализ
2.3.6 Другие методы
ГЛАВА 3. РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
3.1 Кальпаин/кальпастатиновая система у крыс, подвергнутых индуцированной бета-амилоидом нейродегенирации на фоне эстрогенной терапии
3.2 Кальпаин/кальпастатиновая система у крыс, подвергнутых глутамат-индуцированной нейродегенирации на фоне терапии потенциальными нейропротекторами
ВЫВОДЫ
ЗАКЛЮЧЕНИЯ
СПИСОК ЛИТЕРАТУРЫ
ВВЕДЕНИЕ
Нейродегенеративные заболевания 2 болезниАльцгеймера, Паркинсона и другие – представляют в странах с высокой продолжительностью жизни населения огромную медицинскую, социальную, финансовую и научную проблему. Распространенность этих заболеваний растёт по мере старения населения, и большинство развитых стран уже сейчас оказывают специальную поддержку исследованиям в области изучения нейродегенеративных заболеваний.
К настоящему времени участие внутриклеточных протеиназ в нейродегенеративных процессах не вызывает сомнений. С одной стороны, недостаточная интенсивность протеиназозависимых процессов при нейродегенеративных заболеваниях способствует накоплению большого количества патологических белков, например, бета-амилоида и тау-протеина при болезни Альцгеймера, альфа-синуклеина при болезни Паркинсона и других. С другой стороны, протеиназы регулируют основные пути гибели клеток при нейродегенерации – апоптоз, некроз и аутофагию. Таким образом, для нервной ткани жизненно важно поддержание тонкого баланса активности протеиназ, при этом может быть необходимым как повышение их активности для разрушения патогенных белков, так и снижение – для предотвращения массовой гибели нейронов. Са2+-зависимые протеиназы, или кальпаины, относящиеся к семейству С2 цистеинового типа, представлены в тканях ЦНС пятью ферментами – основными μ- и m-кальпаинами (КФ 3.4.22.52 и 3.4.22.53, соответственно) и минорными кальпаинами 3, 5 и 10. Участие Са2+-зависимых протеаз в нейродегенеративных процессах – общебиологическое явление, обнаруживаемое у широкого круга организмов – от нематоды С. elegans до приматов.
В силу особенностей биологии (неспособность к митозу) нейроны особенно чувствительны к накоплению недеградированных продуктов белкового обмена, в норме эффективно разрушаемых внутриклеточными протеиназами. Известно, что в нормальных условиях более 25% синтезированных de novo клеточных белков содержат ошибки и подвергаются быстрому разрушению. Ослабление протеиназоассоциированной функцииконтроля качества клеточных белков (в силу нарушения синтеза и/или регуляции протеиназ из-за сенильных изменений или при действии неблагоприятных экзогенных или эндогенных факторов) приводит к накоплению клеточных белков с ошибками биосинтеза и с нарушенной конформацией. Дефектные белки нефункциональны, участвуют в аберрантных взаимодействиях, образуют нерастворимые белковые агрегаты и приобретают свойства цитотоксичности. В связи с этим, проект направлен на изучение роли внутриклеточных протеиназ в механизмах возрастной и патологической нейродегенерации.
Целью экспериментальной и теоретической работы было изучение роли внутриклеточных кальцийзависимых протеиназ (кальпаинов) в развитии индуцированных нейродегенеративных нарушений и механизмов торможения кальпаинозависимых этапов нейродегенерации путём введения нейропротекторов. Объект исследования - лабораторные крысы- биомодели нейродегенеративных патологий человека. Для достижения поставленной цели было необходимо решить следующие задачи:
1) анализ литературных источников по проблеме разнообразия молекулярных форм кальпаинов в тканях млекопитающих, их эндогенных регуляторов и роли кальпаиновой системы в развитии нейродегенеративных процессов;
2) освоение методических приёмов выделения кальцийзависимых протеиназ из тканей животных, разделения их молекулярных форм, определения протеолитической активности, количественной оценки уровня ингибитора кальпастатина;
3) проведение экспериментов с лабораторными животными: индуцирование нейродегенерации путём введения экзогенных веществ, её коррекция потенциальными нейропротекторами, проведение поведенческих тестов, взятие биологического материала (органов крыс) для биохимического анализа;
4) биохимический анализ протеолитической активности разных молекулярных форм кальпаинов и количественная оценка их ингибитора - кальпастатина в нервной ткани крыс разных экспериментальных групп;
5) анализ результатов об участии кальпаинов и их регуляторов в развитии индуцированной нейропатологии и возможности купирования кальпаинозависимых нарушений потенциальными нейропротекторами.
ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ
1.1.Кальпаин / кальпастатиновая протеолитическая система.
Кальпаин / кальпастатиновая протеолитическая система регулирует широкий спектр клеточных процессов. (Gollet al., 2003; Немова 2010). Она представлена во всех тканях млекопитающих основными формами Са2+-зависимых цистеиновых протеиназ – μ- и m-кальпаинами (КФ 3.4.22.52 и 3.4.22.53, соответственно) и их ингибитором – кальпастатином. (Gollet al., 2003; Nixon2003)Эта высокочувствительная и эффективная система из трех основных компонентов (протеиназ, ингибитора и активатора – Са2+) связана взаимной регуляцией ее составляющих. Дисбаланс в этом протеолитическом союзе, наблюдаемый обычно при повышении уровня внутриклеточного Са2+, приводит к нерегулируемой деградации внутриклеточных структур и усилению кальпаинзависимых путей клеточной гибели, в результате чего развивается тканевая патология: чаще – дегенерация (в скелетных мышцах, сетчатке, тканях ЦНС), иногда – избыточная пролиферация (Carragheretal.,2006; Chakraborti etal., 2012) Определение абсолютного количества разных форм кальпаина и кальпастатина в тканях сопряжено с рядом методических трудностей (Gollet al, 2003); физиологическое соотношение μ- и m-кальпаинов в разных отделах ЦНС оценивается как 1:9, а количество кальпастатина достаточно для полного угнетения активности всего пула кальпаинов, поскольку в общих (неразделенных) тканевых экстрактах активность кальпаинов не выявляется (Gollet al, 2003; Nixon 2003).
Кальпастатин – высокоспецифичный белковый ингибитор μ- и m-кальпаинов – представлен в тканях рядом сплайс-вариантов. В составе комплекса с кальпастатином кальпаин утрачивает способность к примембранной транслокации, аутоактивации и проявлению каталитической активности. Молекулярный механизм взаимодействия кальпаинов и кальпастатина зависит от ионов кальция (Са2+) и к настоящему времени описан в деталях (Moldoveanuet al.,2008). Регуляция уровня кальпастатина в растворимой фракции клетки зависит от скоординированного действия cАМР-зависимой протеинкиназы А (PKA), способствующей образованию агрегатов кальпастатина и сокращению количества его молекул, доступных для взаимодействия с кальпаином, и протеинфосфатазы, индуцирующей освобождение молекул кальпастатина из агрегатов [9]. Кроме того, при длительном повышении активности кальпаинов кальпастатин подвергается кальпаинзависимой деградации, приводящей к еще более значительной активации фермента посредством положительной обратной связи. Вместе с тем, при истощении пула ингибитора стимулируется экспрессия его гена, CAST [9].
Другой внутриклеточный регулятор кальпаинов – ионизированный кальций; по чувствительности к нему различают μ- и m-формы фермента: оптимум для активации μ-кальпаина лежит в микромолярном, а m-кальпаина – в миллимолярном диапазоне Са2+ [10]. Механизм связывания Са2+ и Са2+-индуцируемой активации кальпаинов уже хорошо изучен [11]. Баланс внутриклеточного Са2+ строго регулируется системой Са2+-переносчиков в плазмалемме и мембранах органелл [12]. Однако, развитие тканевой патологии, включая нейродегенерацию, приводит к повышению уровня Са2+ и к ожидаемой активации кальпаинов [4, 7, 13-16]. Поскольку ряд белков-переносчиков Са2+ являются субстратами кальпаинов, в этих условиях происходит их избыточная деградация, еще более усиливающая проблему дисбаланса Са2+ и нерегулируемой активности кальпаинов. Так, при нейродегенерации наблюдается кальпаининдуцированная дисфункция Na+/Ca2+-обменника (NCX3) [17] и потенциалуправляемых Са2+-каналов L-типа [18].
Активация кальпаинов рассматривается как маркер развития нейродегенеративного процесса, естественного или индуцированного в эксперименте. Усиленный гидролиз αII-спектрина кальпаинами, приводящий к появлению двух уникальных фрагментов его расщепления (SBDP150 и SBDP145), является ранним событием в патологии нервной клетки. По образному выражению (Czogalla, Sikorski, 2005), в этом процессе спектрин является мишенью, а кальпаин – снайпером.
Основной формой кальпаинов в миелиновой оболочке является m-кальпаин. Кристаллические структуры m- кальпаинов крысы (Hosfieldet al., 1999) и человека (Stroblet al.,2000) экспрессированных E. Coli(Hosfiend etal.,1999), были установлены методом рентгеноструктурного анализа (рисунок 1).
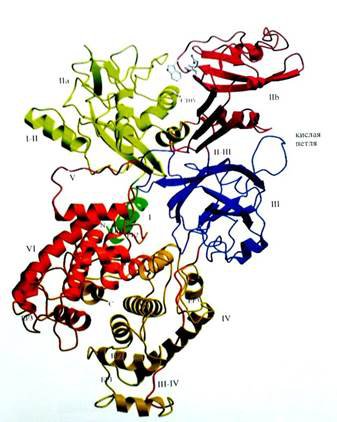
Рис.1 Кристаллическая структура m-кальпаина человека. Домены показаны разными цветами и помечены: I, IIa, IIb, III, IV, V и VI. I-II-α-спираль, связывающая домены Iи IIa. Линкерный домен выделен красной линией III-IV, идущей от промежутка между доменами IIIи IV к правой части рисунка. Указано положение EF-1, EF-2 и EF-3 мотивов в доменах IV и VI. Cys- 105 активного центра, His-262, иTrp-288 на вершине домена IIb отмечены серым цветом (адаптировано из : Reveret. al., 2001a)
1.2.Биохимические механизмы нейродегенеративных заболеваний
Развитие нейродегенеративных заболеваний (НДЗ) связывают, прежде всего, с аккумуляцией патогенных конформеров белков (специфичных для каждого НДЗ) в определенной зоне мозга [19]. Так, в основе патогенеза болезни Альцгеймера (БА) лежит аккумуляция пептидов Аβ1-40 и Аβ1-42 – фрагментов амилоидного белка-предшественника [20], и тау-протеина [21], главным образом, в гиппокампе и коре больших полушарий мозга. Вне зависимости от природы белка с проагрегантными свойствами и локализации белковых агрегатов (вне- или внутриклеточной), всем им присуща цитотоксичность, реализуемая путем изменения базовых биохимических параметров в поврежденных зонах мозга, преимущественно, баланса внутриклеточного кальция [15, 22-24].
Свой вклад в повреждение ЦНС и в дизрегуляцию Са2+ при острых и хронических НДЗ вносит феномен эксайтотоксичности – гиперстимуляции глутаматергических рецепторов, преимущественно рецепторов N-метил-D-аспартата (NMDA), которые являются Ca2+-каналами [24, 25]. Результирующий избыток Са2+ в цитоплазме вызывает персистентную “патологическую” активацию кальпаинов. Нерегулируемый гидролиз субстратов кальпаинов – структурных и регуляторных белков нервной ткани, в том числе проапоптотических, негативно сказывается на морфологии и функциях нейронов и приводит к подавлению базовых механизмов их выживания [14, 15, 26, 27]. Механизмы плейотропного действия кальпаинов в стимуляции нейрональной гибели рассмотрены в недавних обзорах [14, 28].
Предполагают, что изменения активности кальпаинов также могут влиять на секрецию β-амилоидных пептидов, вероятно, через активацию кальпаинами программируемой клеточной смерти – ПКС (Nixonand Mohan, 1999). Однако механизм такого возможного косвенного влияния кальпаинов на β-амилоидную секрецию еще не установлен. Достоверно показано, что кальпаины при болезни Альцгеймера расщепляют белок р35, отвечающий за нормальное развитие нервной ткани, до 25 кДа формы (р25), аккумулирующейся в мозге больных БА (Lee et al., 2000). Активированная 25 кДа форма киназы вызывает пролонгированную активацию циклин-зависимой киназы 5 (Cdk 5) (Kusakawa et al., 2000); в свою очередь, Cdk 5 отвечает за гиперфосфорилирование тау-протеина. Таким образом, р25 (продукт гидролиза кальпаинами) приводит, в конечном итоге, к гиперфосфорилированию тау-протеина и его аккумуляции в неупорядоченных внутриклеточных нейрофиламентах, которая регистрируется у пациентов с болезнью Альцгеймера (Selkoe, 2001). Гиперфосфорилирование придает тау-протеину, обычно легко расщепляемому кальпаинами (Guttmannand Johnsonet al., 1999), высокую устойчивость к деградации μ-кальпаином (Literskyand Johnson, 1992), так что неупорядоченные нейрофиламенты устойчивы к расщеплению кальпаинами. Одной из установленных физиологических функций АРР является ингибирование активности Cdk5, осуществляемое внеклеточным N-концевым доменом АРР (Han et al., 2005). Вследствие этого, в нейронах дефицитных по АРР мышей снижен метаболизм, повышен уровень фосфорилирования тау-протеина, а также они более чувствительны к экзайтотоксическому глутамат-индуцированному апоптозу, механизм которого основан на активации Cdk5. Кроме того, поскольку белки нейрофиламентов являются превосходными субстратами кальпаинов, кальпаины могут играть важную роль в некротической гибели нейронов, которая сопровождает заболевание (Goll et al., 2003).
Предполагаемая роль кальпаинов при болезни Альцгеймера (БА) более комплексна, чем просто нерегулируемая или повышенная активность кальпаинов в результате нарушения гомеостаза Са2+.
С другой стороны, в мозге пациентов повышена концентрация Са2+ (Leissring et al., 2000). Иммуногистохимические исследования показывают, что у страдающих БА повышен уровень m-кальпаина (Grynspan et al., 1997; Tsuji et al., 1998), а также усилен аутолиз µ-кальпаина до 76 и 78 кДа фрагментов (Saito et al., 1993; Marcurn et al., 2005). Это различие аутокаталитической активности в мозге больных и здоровых людей сглаживается добавлением дитиотреитола (ДТТ) (Marcum et al., 2005). Показано, что на фоне 3-кратного повышения интенсивности протекания автолиза кальпаинов (по данным Вестерн-блот анализа), в мозге больных БА наблюдается значительное снижение Са2+-зависимой казеинолитической активности. Объяснением феномена может служить обнаруженное в гиппокампе мозга пациентов, страдающих БА, значительное повышение интенсивности окислительных процессов. Одна из наиболее чувствительных к окислению мишеней – тиоловые группы, составляющие активных центров цистеиновых протеаз. Несмотря на то, что в мозге больных БА и кальпаины, и каспазы находятся в активной конформации, проявлению их протеолитической активности, как предполагают, может препятствовать сильный окислительный стресс, повреждающий наиболее чувствительные зоны мозга. Посмертная казеинолитическая активность в мозге здоровых людей и больных БА одинакова в отсутствие экзогенных восстанавливающих агентов, но в их присутствии (например, ДТТ) у больных БА эта активность значительно выше (Marcurn et al., 2005). Это указывает на то, что в мозге больных значительная доля молекул фермента находится в окисленном состоянии, и их латентная протеолитическая активность в этом случае может проявиться при действии антиоксидантов.
Фармакологические препараты, избирательно воздействующие на кальпаины, в клинике пока отсутствуют. Вместе с тем, среди одобренных к применению препаратов присутствуют регуляторы кальциевых каналов и ряд стероидных гормонов, которые способны влиять на протеолитические каскады и оказывать непрямое модулирующее действие на активность кальпаинов.
ГЛАВА 2.МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
2.1 Реагенты и приборы
В работе были использованы соли, химические реагенты, ингибиторы протеиназ, белковые субстраты, маркеры молекулярных масс, очищенный кальпаин 1 человека, красители, 17β-эстрадиол, L-глутаминовая кислота, нифедипин («Sigma-Aldrich», США), компоненты для вестерн-блот анализа («Bio-Rad», США), поликлональные антитела к кальпастатину sc-20779 и конъюгированные с пероксидазой антитела к IgG кролика sc-2004 («Santa Cruz Biotechnology», США). Предподготовка пептида Aβ1-40 («Sigma-Aldrich», США) включала его растворение в стерильном 0.9% NaCl до конечной концентрации 2.5 г/л и инкубацию раствора при 37 °С в течение недели для агрегации пептида.
В работе были использованы приборы: микроцентрифуга 5417R («Eppendorf», Германия), ультрацентрифуга Optima Beckman LE 80 («Beckman-Coulter», США), твердотельный термостат CH-100 («BioSan», Латвия), камера для электрофореза Mini Protean-3 Cell, камера для блот-переноса Trans-Blot SD Semi-Dry Electrophoretic Transfer Cell, система гель-документирования Gel Doc XR Plus («Bio-Rad», США), спектрофотометр СФ-2000 (ЗАО «ОКБ-Спектр», Россия).
2.2 Моделирование нейродегенерации у лабораторных животных
Исследование проведено на одно- (серии эксперимента I и II) и двухлетних (серия III) крысах Вистар, самцы массой 300 ± 30 г, самки – 250 ± 30 г. Животных содержали в стандартных виварных условиях при естественном световом режиме и свободном доступе к воде и пище. Способы воздействия на животных в трех сериях эксперимента обобщены в таблице 1.
В каждой серии эксперимента произвольно выделяли четыре группы животных (в каждой n = 7-10): 1) интактные; 2) контрольные – ложно-оперированные; 3) подвергнутые введению нейротоксичного агента; 4) подвергнутые введению нейротоксичного агента на фоне введения потенциального нейропротектора.
| группа крыс, воздействие | серия эксперимента | ||
| I | II | III | |
| 1. интактные | + | + | + |
| 2. ложно-оперированные | NaCl | NaCl | NaCl |
| 3. нейротоксичный агент | Аβ1-40 | глутамат | глутамат |
| 4. нейротоксичный агент + потенциальный нейропротектор | Аβ1-40 + 17β-эстрадиол | глутамат + 17β-эстрадиол | глутамат + нифедипин |
Таблица 1. Схема экспериментов с лабораторными животными. Дозы, длительность и способ введения веществ приведены в тексте.
Ложнооперированные животные (группа животных 2), подвергались однократной инъекции 2 мкл 0.9% раствора NaCl в C1-область правого гиппокампа. Животные групп 3 и 4 подвергались однократной интрацеребральной (в ту же область мозга) инъекции агента, индуцирующего нейродегенеративные изменения: в I серии эксперимента – 2 мкл раствора, содержащего 5 мкг пептида Аβ1-40, во II и III – 2 мкл 1 мкМ раствора L-глутаминовой кислоты. Помимо инъекции нейротоксичного агента животным 4-й группы ежедневно в течение 14 дней с момента операции вводили 0.1 мг 17β-эстрадиола интраназально (I и II серии эксперимента) или 1 мг нифедипина per os (III серия). Интрацеребральная инъекция выполнялась в стерильных условиях под хлоралгидратным наркозом (400 мг/кг веса), забой проводили через две недели после операции декапитацией после аналогичной инъекции хлоралгидрата. Для оценки когнитивной функции крыс до и после оперативного вмешательства проводились поведенческие тесты в открытом лабиринте с параллельными ходами или в водном лабиринте Морриса.
Биохимические и гистоморфологические показатели оценивали в разных отделах головного мозга крыс: поврежденном инъекцией правом гиппокампе и в интактных левом гиппокампе и коре правого и левого больших полушарий.
2.3 Анализ биохимических показателей
2.3.1 Экстракция белков из тканей
У лабораторных животных (крыс) для последующего биохимического анализа были взяты образцы нервной ткани – гиппокампа и коры больших полушарий. Учитывая дисперсность пула кальпаинов в клетке, связанную со спецификой их регуляции, определяли показатели как в цитозольной фракции клеток, так и во фракции мембрано-связанных белков, полученных по методу (Enns, Belcastro, 2006). Цитоплазматическую фракцию получали из ткани, гомогенизированной в 20 мМ Трис-HCl-буфере (рН 7.5) с добавлением 80 мМ КСl, 5 мМ Na-соли этилендиаминтетрауксусной кислоты (EDTA) и 20 мМ дитиотреитола (DTT) (соотношение 1:10, вес/объем) с последующим центрифугированием (20 000 g, 20 мин). Фракцию мембраносвязанных белков – повторным центрифугированием (20 000 g, 20 мин) получившегося осадка, предварительно ресуспендированного в равном объеме того же буфера с добавлением 0.33% тритона X-100. Буфер для гомогенизации включал смесь ингибиторов протеиназ: лейпептина (0.5 мкг/мл), пепстатина (1 мкг/мл), апротинина (1 мкг/мл) и 1 мМ фенилметилсульфонилфторида (PMSF).
Буфер для гомогенизации, (объём 500 мл)
• 20 мМ Трис, навеска 1,2114 г
• 80 мМ хлорида натрия, навеска 2,98 г
• 5 мМ ЭДТА, навеска равна 0,9306 г
• 20 мМ ДТТ, навеска равна 1,54 г
• растворить в дистиллированной воде, довести рН до 7,5 5 конц. HCl
• довести водой до 500 мл в колбе
Буфер для гомогенизации с тритоном Х-100 (объём 100 мл)
• 0,33 % тритон Х-100, навеска 0,33 г.
• довести до 100 мл буфером для гомогенизации.
Ингибитор PMSF (объем 10 мл)
• 100 мМ PMSF, навеска 174 мг
• растворить в 10 мл изопропанола
• разлить по аликвотам 0,5 мл, заморозить при -20 оС
2.3.2 Определение активности кальпаинов
В полученных фракциях цитозольных и мембраносвязанных белков оценивали активность кальпаинов – Са2+-зависимую казеинолитическую активность, чувствительную к ингибиторам цистеиновых протеиназ (Enns, Belcastro, 2006). Реакционная смесь, объемом 500 мкл, включала 0.5 мг казеина, денатурированного щелочью, 20 мМ DTT, 200 мкл ферментной фракции и 2.5 мМ СаСl2или EDTA (для оценки Са2+-зависимой и Са2+-независимой активности, соответственно) в 50 мМ Трис-HCl-буфере (рН 7.5). После 30-минутной инкубации (28°С) в аликвотах 100 мкл спектрофотометрически определяли содержание остаточного белка по методу Брэдфорд (Bradford, 1976). Единицу активности кальпаинов (ед.акт.) определяли как количество фермента, вызывающее увеличение оптического поглощения на 0.1 OE при 595 нм за 1 ч реакции в указанных условиях. Удельную активность определяли в расчете на 1 мг белка соответствующей фракции.
1. Буфер для реакции (объём 500 мл)
• 20 мМ ДТТ, навеска 1.55 г
• 70 мМ Трис, навеска 4,235 г
• прилить дистиллированной воды, довести рН до 7,5 конц. HCl
• довести дистиллированной водой до 500 мл
2. На буфере для реакции – 0,1 М раствор хлорида кальция (объём 100 мл)
• хлорид кальция, навеска 1,47 г
• довести до 100 мл буфером для реакции
3. На буфере для реакции 0,1 М раствор ЭДТА (объём 100 мл)
• ЭДТА, навеска 3,72 г
• довести до 100 мл буфером для реакции
2.3.3 Зимография с козеином
Характеризацию пула кальпаинов проводили также методом зимографии с казеином согласно методу (Arthur, Mykles, 2000). Общую фракцию цитоплазматических и мембраносвязанных белков получали центрифугированием тканевых гомогенатов (105000 g, 30 мин) в 50 мМ HEPES-NaOH-буфере (рН 7.6), содержащем 150 мМ NaCl, 10% глицерин, 0.1% тритон Х-100, 5 мМ EDTA, 10 мМ 2-меркаптоэтанола и ингибиторы протеиназ (0.1 мМ PMSF, 10 мкг/мл лейпептин) (соотношение 1:10, вес/объем). Разделяли белки (30 мкг на дорожку) в 12% ПААГ, сополимеризованном с казеином («Bio-Rad», США) в концентрации 0.2%. Этап нативного электрофореза сопровождался инкубацией геля в 50 мМ Трис-HCl-буфере (рН 7.2), содержащем 10 мМ меркаптоэтанола и 5.0 мМ СаCl2, и его стандартной окраской Coomassiebrilliant blueR-250.
Реактивы для зимографии:
1. Раствор казеина:
10 мг/мл казеина в 0,75 М Трис-HCl-буфере, рН=8,8 (50%-ный буфер А).
2. Буфер для инкубации с кальцием (объем 1 л):
· 50 мМ Трис-HCl-буфер, навеска 6,057 г, рН = 7-7,2 при комнатной температуре
· 5 мМ хлорид кальция, навеска 0,735 г
· 10 мМ ДТТ, навеска 1,54 г
· Довести объем в колбе до 1 л б/д водой
2.3.4 Электрофорез белков в полиакриламидном геле
Электрофорез белков в полиакриламидном геле – метод разделения смесей белков в полиакриламидном геле в соответствии с их электрофоретической подвижностью, детерминированной длиной полипептидной цепочки, молекулярной массой, укладкой белковой молекулы, посттрансляционными модификациями и другими факторами. Данный способ фракционирования белков и пептидов широко применяют в современной молекулярной биологии, биохимии, генетике.
Разработано большое количество модификаций электрофореза белков в полиакриламидном геле для решения разных задач и для различных белков и пептидов. Наиболее распространённой разновидностью является электрофорез белков в полиакриламидном геле (ПААГ) в присутствии додецилсульфата натрия (SDS) по Лэммли (Laemmli, 1970).
1. 30 % раствор акриламида (акриламид : бис- акриламид =74,1 : 1) (объем 100 мл)
· 29,6 г акриламида;
· 0,4 г бис-акриламида;
· растворить в 100 мл б/д воды;
· профильтровать; хранить в тёмном месте.
2. Буфер А: 1,5 М трис-HCl-буфер, рH =8,8 (объем 200 мл)
· 36,312 г трис на 200 мл б/д воды
3. Буфер В: 0,5 М Трис-НCl-буфер, pH= 6,8 (объем 200 мл)
· 12,114 г трис на 200 мл б/д воды
· рН довести концентрированной HCl до нужного рН
4. 50% SLB-буфер:
· 20% глицерин, берём для приготовления 25 мл глицерина
· 25 мл 0,1 М Трис-HCl, рН=6,8
· 10 мМ ЭДТА, навеска 0,47 г
· 10 мМ 2-меркаптоэтанола, берём для приготовления 100 мкл
· 0,02 % бромфенолового синего, навеска 0,03 г
5. Буфер для электрофореза (5-кратный), рН = 8. Не требует подведения рН (объем 1 л):
· 125 мМ Трис, навеска 15,14 г
· 626 мМ глицерина, навеска 46,92 г
· 5 мМ ЭДТА, навеска 1,86 г.
· Непосредственно перед использованием разбавить до однократного, добавить 10 мМ 2-МЭ (или 1 мМ ДТТ), для этого к 200 мл 5х-буфера прилить 800 мл б/д воды и добавить 0,8 мл 2-МЭ
6. Фиксаж:
· 45% этанол
· 45% вода
· 10% уксусная кислота
7. Окраска:
· 0,5% кумасси
· 10% уксусная кислота
· 40% этанол
· 50% вода
8. Отмывка:
· 10% уксусная кислота
9. Лизирующий буфер (для гомогенизации) (объём 200 мл)
· 50 мМ HEPES, навеска 2,384 г, pH7,6 (довести конц. щёлочью)
· 150 мМ хлорид натрия, навеска 1,77 г
· 10% глицерин, объём 20 мл
· 0,1% Тритон Х-100, объём 0,2 мл
· 5 мМ ЭДТА, навеска 0,3724 г.
· 10 мМ ДТТ, навеска 0,308 г.
2.3.5 Вестерн-блот анализ
Вестерн-блот анализ по тестированию количества кальпастатина включал разделение тканевых белков (30 мкг на дорожку) методом электрофореза в 12% ПААГ в присутствии SDS (Laemmli, 1970) с последующим полусухим переносом полипептидов на нитроцеллюлозную мембрану (буфер: 48 мМ Трис-HCl, 39 мМ глицин, 0.0375% SDS, 20% метанол, pH 9.2). После инкубации (2 ч, 20°C) мембраны в буфере TBS(50 мМ Трис-HCl-буфер, 150 мМ NaCl, pH 7.5) проводили блокировку сайтов неспецифического связывания 5% раствором обезжиренного молока в буфере TBST(TBS с добавлением 0.1% Tween 20, pH 7.5) в течение 1 ч. Далее мембрану подвергали последовательному экспонированию с поликлональными антителами к кальпастатину (разведение 1 : 2500 в буфере TBST; 1 ч) и с антителами к IgG кролика, конъюгированными с пероксидазой (разведение1 : 2000 в буфере TBST; 1 ч); каждый из указанных этапов завершался многократной отмывкой буфером TBST. Мембрану подвергали стандартной обработке системой Immune-Star(Bio-Rad, США).
2.3.6 Другие методы
Концентрацию белкаво фракциях определяли методом Брэдфорд (Bradford, 1976) с использованием бычьего сывороточного альбумина (BSA) в качестве стандарта.
Денситометрия полос на зимограммах и рентгеновской пленке проводилась с помощью стандартной программы “Image J”.
Статистическую обработку данныхпроводили с применением общепринятых методов вариационной статистики с использованием пакетов программ MSExcel и StatGraphics. Достоверность различий оценивали с помощью непараметрического критерия U (критерий Вилкоксона-Манна-Уитни), а также с использованием однофакторного дисперсионного анализа (Коросов, Горбач, 2007).
ГЛАВА 3.РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
3.1. Кальпаин/кальпастатиновая система у крыс, подвергнутых индуцированной бета-амилоидом нейродегенерации на фоне эстрогенной терапии
Нейродегенерация индуцировалась у крыс линии Вистар старших возрастных групп – 12 и 24 месяцев. Экспериментальное воздействие заключалось в интрацеребральном введении 42-членного фрагмента амилоидного белка-предшественника – Абета(1-42) (экспериментальная модель болезни Альцгеймера), а также сочетанном интрацеребральном введениие бета-амилоидного пептида и интраназальном введении нейропротектора (эстрадиола). Среди животных были выделены: группа контроля – ложно-оперированные (2 μл физраствора в область правого гиппокампа); первая опытная группа – 2 μл раствора пептида Абета(1-42) (соответствуют 5μг пептида) в область правого гиппокампа; вторая опытная группа – после аналогичной инъекции пептида Абета(1-42) ежедневное интраназальное введение 0,1 мг 17-бета-эстрадиола.
Было обнаружено, что в присутствии амилоидогенного пептида в нервной ткани происходит активация кальпаиновой системы, причем степень активации положительно коррелирует с интенсивностью гибели клеток нервной ткани (плотностью нейронов). Регуляция кальпаинов может осуществляться как на уровне синтеза ферментного белка (или отдельных его форм – продуктов разных генов), так и на посттрансляционном уровне за счет процессов аутолиза, связывания с эндогенным ингибитором кальпастатином или с аллостерическим регулятором – кальцием.
Обнаруженное методом казеиновой зимографии увеличение пула аутолизированных кальпаинов (118 кДа) в группе животных №2 отражает активацию кальпаинов in vivo. Наблюдаемая активация m-кальпаина (120 кДа), по-видимому, как и во многих других ситуациях, сопряжена с избытком кальция в цитоплазме. Еще одним подтверждением этого пути регуляции активности кальпаинов служит стабильный уровень их ингибитора – кальпастатина, обнаруженный в нашем исследовании.
Как оказалось, терапия эстрадиолом обращает эффект гиперактивации кальпаинов, при этом снижается как общая активность кальпаинов в нервной ткани, так и активность индивидуальных фракций. В присутствии эстрадиола меньшая доля предшественника кальпаина подвергается аутолизу, а следовательно, активации. Необходимы дальнейшие исследования механизма регуляции активности кальпаинов у экспериментальных животных, в частности, необходимы эксперименты, направленные на установление источника избыточного кальция и поиск средств, предотвращающих эти патологические токи.
Уровень синтеза протеасом в мозговой ткани невысок в сравнении с другими органами и имеет тенденцию к снижению с возрастом (при сравнении показателей у 18, 24 и 30-месячных животных), о чем судили по количеству альфа-1,2,3,5,6,7 субъединиц протеасом, формирующих альфа-кольца коровой 20S частицы, универсальной для 20S и 26S протеасом.
Было подтверждено изменение уровня экспрессии и активности катепсинов в разных зонах мозга у экспериментальных животных. Интрацеребральное введение бета-амилоидного пептида привело в нашем эксперименте к значительному повышению (p<0,05) экспрессии гена катепсина D в коре (правом полушарии) головного мозга крыс по сравнению с животными контрольной группы. В неокортексе крыс, которые после введения бета-амилоидного пептида получали эстрадиол, относительный уровень экспрессии данного гена не отличался от контрольных значений, то есть восстанавливался до нормального уровня. В области правого гиппокампа введение бета-амилоидного пептида привело к увеличению экспрессии гена катепсина D приблизительно в 7 раз (p<0,001). Последующее введение эстрадиола привело к еще большему возрастанию (в 30 раз) относительной экспрессии указанного гена (p<0,001).
Наши данные продемонстрировали значительное влияние бета-амилоида и эстрадиола на когнитивные функции крыс, оцененные с помощью водного лабиринта Морриса. У заранее обученных самок и самцов крыс после введения бета-амилоидного пептида в гиппокамп значительно (p<0,05) увеличилось время поиска скрытой подводной платформы. Последующее введение эстрадиола сокращало время поиска подводной платформы у животных обоих полов по сравнению с таковыми, получившими только бета-амилоид. При этом у самок время поиска уменьшилось более значимо (p<0,01), чем у самцов (p<0,05).
Результаты конфокальной микроскопии срезов мозга показали, что введение бета-амилоидного пептида привело к значительному увеличению уровня его иммунореактивности в тканях как правого, так и левого (в меньшей степени) полушария головного мозга. Эти результаты, наряду с поведенческими данными, свидетельствуют об эффективности введенного препарата амилоидного пептида и служат доказательством репрезентативности выбранной модели болезни Альцгеймера. Последующее введение эстрадиола привело к снижению количества бета-амилоида в мозге крыс почти до контрольного уровня. Сокращение амилоидных депозитов у крыс, получавших эстрадиол, свидетельствует о запуске в нервной ткани неких адаптационных процессов, направленных на их утилизацию.
Механизм нейропротекторной роли эстрадиола пока полностью не понят, хотя отмечен этот феномен давно. Использование эстрадиола в качестве нейропротектора продиктовано несколькими причинами. Во-первых, нейропротективный эффект эстрадиола довольно хорошо известен и описан в литературе (McEwen et al., 2001; Asimiadou et al., 2005; Lebesgue et al., 2009). Во-вторых, показано, что эстрадиол в полном смысле слова является нейростероидом, так как в мозге имеются все ферменты, необходимые для его синтеза (Stoffel-Wagner, 2001; Reddy, 2010). В-третьих, известно, что нейропротективное действие эстрадиола связано с его антиоксидантным (Behl et al., 1997) и антиапоптотическим (Asimiadou et al., 2005) действием, то есть с эффектами, противоположными эффектам пептида Aбета.
Полученные результаты могут свидетельствовать об адаптивной реакции нервной ткани на введение токсичного пептида. В некоторых публикациях указывается на то, что система лизосомальной аутофагии может принимать участие в деградации бета-амилоидного пептида (Nixon, 2007). Вероятно, эстрадиол стимулирует аутофагию белкового материала; об этом свидетельствуют как данные о содержании пептида Aбета в мозговой ткани, так и оценка активности и уровня экспрессии протеиназ лизосом. Методом флюоресцентной иммуногистохимии было установлено снижение количества бета-пептида у крыс, получавших нейропротективную терапию эстрадиолом.
На основании данных, полученных в эксперименте можно сделать следующие выводы. 1. Уровень экспрессии генов лизосомальных протеиназ CtsD, CtsB, CtsL и альфа-субъединиц протеасом и активность кодируемых ими ферментов в коре головного мозга крыс при старении снижается. Напротив, активность кальпаинов у животных старших возрастных групп повышается. 2. Уровень экспрессии и активности катепсина D, а также интенсивность кальций-зависимого протеолиза значительно возрастают в гиппокампе и коре головного мозга крыс после интрацеребрального введения бета-амилоидного пептида, при этом страдает когнитивная функция животных. 3. Введение эстрадиола на фоне бета-амилоидной интоксикации приводит к уменьшению содержания этого пептида в гиппокампе крыс и улучшению биохимических и поведенческих показателей у экспериментальных животных. 4. Фармакологическая активация лизосомальной функции эстрогенами может способствовать удалению Aбета при болезни Альцгеймера; нормализация кальциевого гомеостаза в этой ситуации предотвращает патологическую активацию кальпаиновой системы, а, вместе с тем, и потерю нейронов по кальпаин-зависимым путям клеточной гибели.
3.2 Кальпаин/кальпастатиновая система у крыс, подвергнутых глутамат-индуцированной нейродегенерации на фоне терапии потенциальными нейропротекторами
Для индуцирования патологической нейродегенерации нами была использована модель глутаматной токсичности, сопровождающей многие нейродегенеративные заболевания и нормальное старение. Глутамат – самый распространенный в центральной нервной системе возбуждающий нейромедиатор; около 40% всех синапсов головного мозга – глутаматергические (Fairman, Amara, 1999). Хотя глутамат является жизненно важным нейромедиатором, он также является и мощным нейротоксином, который может вызывать гибель нервных клеток (Kim et al., 2011). Глутамат освобождается из глутаматергических нервных окончаний в синаптические щели и в норме должен быть удален из них (Kanai, Hediger, 2003). Накопление избытка внеклеточного глутамата и последующая гиперстимуляция глутаматергических рецепторов приводит к увеличению продукции активных форм кислорода и азота, которые индуцируют оксидативный стресс и гибель нейронов (Ganel, Rothstein, 1999; Kim et al., 2011). Предполагается, что основной вклад в эксайтотоксичность вносит активация NMDA-рецепторов (Tanovic, Alfaro, 2006), которые являются Ca2+-каналами. В соответствии с ранее поставленными задачами, были получены следующие результаты:
1. Проявления нейродегенерации, индуцированной острым воздействием экзогенного глутамата, о которых судили по результатам гистоморфологии нервной ткани различных зон головного мозга (правого и левого полушарий, гиппокампа) и поведенческих тестов, менее выражены в сравнении с нейродегенерацией, индуцированной амилоидным бета-пептидом (. Нами не выявлено существенного повреждения тканей центральной нервной системы при использованной дозе эксайтотоксического агента, что подтверждается как микроскопическими, так и молекулярно-биологическими данными, вместе с тем, страдает когнитивная функция животных. Проведенное тестирование поведенческих реакций (водный лабиринт Морриса) позволило оценить степень нарушения когнитивной функции у экспериментальных животных. Так, в группе 3 (острое воздействие глутамата) нарушения были оценены как умеренные, а между группами животных 1, 2, 4 различия отсутствовали (нарушений не наблюдалось). Таким образом, в сравнении с проявлениями амилоидной токсичности нейротоксичность глутамата имеет менее выраженный характер.
2. Установлено, что глутаматная токсичность оказывает специфическое действие на характер ответной реакции различных протеолитических систем клетки и, как следствие, регулируемых ими процессов клеточной гибели и выживания. На изученной нами модели нейродегенерации показано, что уровень экспрессии генов лизосомальных протеиназ (катепсинов В, D и L), конститутивных субъединиц протеасомы, а также активность кодируемых ими ферментов достоверно не изменяются в гиппокампе и коре головного мозга крыс после острого воздействия глутамата, тогда как снижается интенсивность кальций-зависимого протеолиза.
Интрацеребральное введение глутамата привело к значительному (до 50%) снижению протеолитической активности кальпаинов, при этом тормозился и процесс их аутокаталитической активации, и активность полноразмерной и аутолизированной форм. Уровень ферментов и их внутриклеточного ингибитора, кальпастатина, при этом не изменялся, что свидетельствует о регуляторных воздействиях, по всей видимости, о снижении уровня внутриклеточного кальция. Наши результаты, в первом приближении, противоречат установленному учеными из мединститута Г.Хьюза в 2000 году механизму участия кальпаинов в нейродегенерации (Lee et al., 2000), который со временем находит все больше экспериментальных подтверждений и, по-видимому, в общих чертах универсален для большинства нейродегенеративных заболеваний. Согласно классическим представлениям, цитотоксический эффект агентов разной природы (глутамата, амилоидного бета-пептида и других белковых агрегатов), сопряжен с нарушением кальциевого гомеостаза (LaFerla, 2002), а избыток Са2+ в цитоплазме вызывает персистентную «патологическую» активацию кальпаинов (Goll et al., 2003; Araújo et al., 2007). Нерегулируемый гидролиз субстратов кальпаинов – структурных и регуляторных белков нервной ткани, в том числе проапоптотических – негативно сказывается на морфологии и функциях нейронов и приводит к подавлению базовых механизмов их выживания (Bezprozvanny, 2009; Vaisid et al., 2008; Vosler et al., 2008). Таким образом, роль кальпаинов в нейродегенерации реализуется на многих этапах – от образования токсичных конформеров клеточных белков в ходе их «патологического» процессинга кальпаинами до реализации программ клеточной гибели по путям апоптоза и(или) некроза. Однако, в последнее десятилетие все больше доказательств находит гипотеза о том, что вышеописанная последовательность событий в развитии кальпаин-зависимой нейродегенерации – лишь вторичное явление, а на ранних этапах нейропатологии имеет место дефицит кальция и кальций-регулируемых процессов, включая функциональную активность кальпаинов (Chen et al., 2001). По-видимому, этот этап кратковременен и протекает в отсутствие видимых признаков заболевания, но, вполне вероятно, на модели острой глутаматной токсичности мы смогли уловить именно эту фазу биохимических изменений в клетках; отдаленные последствия мы смогли бы проследить только в том случае, если бы перешли к модели хронической глутаматной токсичности, например, путем интрацеребрального микродиализа.
Полученные нами результаты свидетельствуют о том, что экспрессия гена лизосомальной протеиназы катепсина D CtsD у крыс, которым вводили глутамат в правый гиппокамп, практически не изменилась в изученных отделах мозга у самок крыс, но оказалась снижена у самцов. Так был получен неожиданный для нас результат, свидетельствующий о вероятном блокирующем эффекте глутамата на индукцию экспрессии катепсина D. Наши данные указывают на то, что у самок крыс введение глутамата не оказывало столь значительного повреждения в правом полушарии головного мозга, как у самцов. Полученные нами данные о половой специфике ответной реакции хорошо согласуются с данными литературы: исследования на крысах и мышах показывают существование половых различий в количестве погибших нейронов после фокального ишемического повреждения (окклюзия средней мозговой артерии).
3. Показано, что эстрадиол (потенциальный нейропротектор) оказывает влияние на уровень синтеза и протеолитической активности лизосомальных протеиназ (но не кальпаинов), а его введение животным, подвергнутым острому воздействию глутамата, приводит к активации протеиназ, отвечающих за аутофагию (лизосомальных катепсинов), и к восстановлению физиологического уровня активности кальпаиновой системы в мозге крыс, а также к улучшению других биохимических и поведенческих показателей у экспериментальных животных. Воздействие эстрадиола оказалось положительным при разных типах нейродегенерации (амилоидной и глутаматной).
Индивидуальное введение эстрадиола (группа 2, ложно-оперированные + эстрадиол) отразилось на уровне экспрессии гена CtsD (но не генов кальпаинов Capn1, Capn2) у животных, причем по-разному у самок и самцов. Так, у самцов экспрессия гена CtsD в левом гиппокампе увеличилась по сравнению с контролем более чем в 1,5 раза. Такое изменение в левом гиппокампе мы можем объяснить адаптивными механизмами, которые были индуцированы введением эстрогена после повреждения контралатерального гиппокампа введением физиологического раствора. По всей видимости, левый, неповрежденный гиппокамп был вынужден частично взять на себя выполнение функций поврежденной доли гиппокампа, что привело к адаптивному увеличению экспрессии гена катепсина D, о связи которого с когнитивными функциями известно из литературы (Payton et al., 2003; Payton, 2006). Аналогичное изменение у самцов крыс, получавших эстрадиол, было нами обнаружено в экспрессии гена CtsD в коре правого полушария, где экспрессия данного гена в 2,25 раза превышала контрольный уровень (р<0,01). Повышение экспрессии CtsD под влиянием эстрадиола может свидетельствовать об усилении аутофагических процессов в нейронах головного мозга крыс, необходимых, в частности, для получения пула свободных аминокислот и компенсации травматического повреждения. Отсутствие аналогичного эффекта у самок крыс можно объяснить изначально более высоким уровнем нейропротективного гормона эстрадиола в кровеносной системе и мозге. Увеличение экспрессии гена катепсина D под влиянием эстрадиола может быть одним из механизмов, позволяющим осуществлять адаптивные перестройки в нервной системе и способствующим пластическим изменениям в ней при повреждениях.
4. Установлено, что эффективность эстрогенной терапии при данном типе нейропатологии значительно выше у самок, чем у самцов. У самок крыс с экспериментальной неродегенерацией введение эстрадиола (группа 4) приводило к восстановлению утраченной активности кальпаинов до нормального уровня, обнаруженного у крыс групп 1 и 2; у самцов действие эстрадиола на фоне глутаматной интоксикации не было выявлено. У самок не наблюдались изменения экспрессии гена CtsD ни при изолированном введении глутамата (группа 3), ни при введении эстрадиола (группа 2). Однако совместное введение глутамата и эстрадиола (группа 4) привело к увеличению экспрессии гена катепсина D в коре правого полушария самок, где она была увеличена по сравнению с контролем приблизительно в 2,1 раза (р<0,01). Значительное повышение экспрессии катепсина в данном случае, по нашему мнению, следует считать адаптивной реакцией на повреждение, которая не была полностью подавлена глутаматом. Известно, что экспрессия гена катепсина D находится под контролем эстрогенов. Так, известно о жесткой регуляции экспрессии катепсина D в клетках эстроген-зависимых опухолей молочной железы (Rochefort et al., 1998; Cavaillès et al., 1993). Имеются также сведения о том, что эстрогены стимулируют транскрипцию гена катепсина D через элементы эстрогенового ответа, расположенные вблизи промоторной области гена (Cavailles et al., 1991).
У самок, в отличие от самцов, изменение экспрессии гена CtsD было отмечено только в группе 4, которой вводили и глутамат, и эстрадиол. Поскольку ни в одном случае, когда вводили один лишь глутамат, изменений экспрессии не наблюдалось, можно предположить, что в группе 4 изменение экспрессии гена CtsD было связано с введением эстрадиола и опосредовано эстрогеновыми рецепторами. При этом повышение экспрессии катепсина D в этом случае, по всей видимости, не может быть связано с аутофагической клеточной смертью. Различную реакцию на введение эстрадиола у самок и самцов крыс (у самок изменения выявлены только в правом неокортексе, тогда как у самцов – ещё и в левом гиппокампе) можно объяснить различным распределением эстрогеновых рецепторов в мозге крыс разного пола, а также их различной чувствительностью к нейрональному повреждению.
Мозг самок в значительной степени защищен от клеточной смерти (Alkayed et al., 1998; Rusa et al., 1999; Simpkins et al., 1997). Введение невысоких доз эстрадиола предотвращает апоптоз в коре мозга в области пенумбры как у самок, так и самцов крыс (Dubal et al., 1998; Rusa et al., 1999; Toung et al., 1998). В данной модели мозгового повреждения нейропротективная активность эстрадиола зависит от наличия рецепторов ER-альфа (Dubal et al., 2001; Rau et al., 2001). Экспрессия мРНК ER-альфа очень низка в коре мозга взрослых особей, однако после унилатерального ишемического повреждения уровень транскриптов гена ER-альфа в коре быстро повышался на поврежденной стороне мозга (Dubal et al., 2006). Активация гена ER-альфа не зависела от введения эстрадиола, хотя имелись некоторые различия в динамике увеличения экспрессии. Кроме того, повышение экспрессии гена ER-альфа является необходимым для нейропротективного эффекта эстрадиола, поскольку у самок мышей, нокаутированных по гену ER-альфа, эстрадиол не оказывал протективного воздействия (Dubal et al., 1999). У самцов мышей с фокальной ишемией мозга наблюдалась гораздо более значительная потеря нервных клеток, чем у самок, хотя нейропротективный эффект эстрадиола также отмечался (Alkayed et al., 1998; Toung et al., 1998).
Таким образом, нами выявлен повышенный уровень экспрессии гена CtsD в коре правого полушария и в левом гиппокампе самцов крыс при введении эстрадиола, а также значительное повышение экспрессии этого гена в коре правого полушария самок крыс при введении эстрадиола на фоне эксайтотоксического повреждения. Показано, что эстрадиол защищает гиппокамп и кору мозга от избыточной гибели нейронов при эксайтотоксическом и травматическом повреждении. Нейропротективный эффект эстрадиола зависит от пола животных; у самок он более выражен.
5. Установлено, что одной из составляющих нейропротективного действия эстрадиола, помимо описанных ранее антиоксидантного и антиапоптотического действия, является активация лизосомального протеолиза (катепсинов) и стабилизации активности кальпаиновой системы, которая достигается, по-видимому, за счет нормализации кальциевого гомеостаза в тканях мозга. Многочисленные научные данные подтверждают гипотезу о нейропротективной роли аутофагии и указывают на то, что патогенные и аберрантные белки могут подвергаться секвестрированию и, возможно, разрушению в аутофагических вакуолях (Terman et al., 1999; Cuervo, Dice, 2000; Terman, Brunk, 2004; McCray, Taylor, 2008; Nixon, Yang, 2011). Так, данные иммуноэлектронной микроскопии свидетельствуют о солокализации катепсина D с амилоидными бляшками в коре головного мозга у пациентов с болезнью Альцгеймера (Cataldo, Nixon, 1990), а нами установлен факт их солокализации со скоплениями амилоида в мозге крыс – моделей болезни Альцгеймера (результаты первого этапа; Рендаков и др., в печати). В норме катепсины являются внутриклеточными протеолитическими ферментами, ассоциированными с лизосомами; однако обработка срезов мозга антителами к катепсину B и катепсину D показала, что высокая иммунореактивность этих ферментов обнаруживается также в амилоидных бляшках у модельных животных, а также сенильных амилоидных бляшках у человека. При этом внеклеточные сайты с катепсиновой иммунореактивностью не обнаруживаются в мозге контрольных пациентов соответствующего возраста, не имеющих неврологических заболеваний, также как и в мозге пациентов с болезнью Хантингтона или Паркинсона (Cataldo, Nixon, 1990).
Фармакологические препараты, избирательно воздействующие на протеолитические пути клеточной гибели и баланс внутриклеточного Са2+, в клинике пока отсутствуют. Вместе с тем, ряд эндогенных веществ, например, половые стероиды, обладают антиоксидантными, антиамилоидными, антиагрегантными свойствами и способны влиять на протеолитические каскады, в связи с чем могут быть перспективны для терапии нейродегенерации.
ВЫВОДЫ
В ходе выполнения дипломной работы выполнен ряд учебно-научных задач.
Проведен анализ литературных источников по исследуемой проблеме.
Освоены некоторые методические приемы выделения, определения активности, идентификации основных молекулярных форм кальций-активируемых протеиназ, количественной оценки их ингибитора кальпастатина в тканях млекопитающих (на примере крыс).
В эксперименте по моделированию болезни Альцгеймера у лабораторных животных (крыс) обнаружены изменения кальпаин / кальпастатиновой системы, направленные на повышение каталитической активности Са2+-зависимых протеиназ и тесно связанные с дисбалансом внутриклеточного Са2+. В связи с этим, результаты изучения предложенной модели нейродегенерации можно экстраполировать на широкий круг родственных нарушений.
Показано, что в тканях мозга (коре больших полушарий, гиппокампе) крыс присутствуют основные, типичные для большинства тканей млекопитающих внутриклеточные протеиназы кальпаины, при этом можно выделить особенности в профиле их экспрессии, энзиматической активности и соотношении молекулярных форм, характерные для различных тканей (тканеспецифичность), а также для здоровых и патологически измененных тканей. Наиболее подробные данные получены для кальций-активируемых протеиназ (кальпаинов), поскольку их роль в развитии нейродегенерации, как возрастной, так и патологической, несомненна, но детально к настоящему времени не изучена.
Анализ биохимических изменений в мозге животных с амилоид-индуцированной нейродегенерацией показал, что ведущую роль в развитии тканевой патологии (избыточной гибели нервных клеток по механизмам апоптоза и некроза) играет дизрегуляция кальпаиновой системы. Нарушения в работе указанной протеолитической системы вызваны, во-первых, снижением содержания в тканях их естественного ингибитора, кальпастатина, то есть нарушением баланса протеиназа / ингибитор, а, во-вторых, нарушением баланса внутриклеточного Са2+, сопровождающим развитие дегенеративных изменений во многих тканях, в том числе, нервной. В модельном эксперименте получены данные, подтверждающие возможность модулирования процесса нейродегенерации за счет введения половых стероидов (эстрадиола), обладающих, наряду с ранее показанным антиоксидантным и антиапоптотическим действием, способностью к избирательной регуляции Са2+-зависимых протеиназ в тканях.
Полученные данные отличаются новизной, дополняют имеющиеся в литературе сведения о протеолитических ферментах млекопитающих. Результаты актуальны как в области фундаментального естествознания для изучения основных компонентов протеолитического аппарата клетки и понимания базовых основ его функционирования в норме и патологии, так и в прикладном аспекте (биомедицина, фармацевтика) поиска подходов к регуляции протеолитических процессов, лежащих в основе патологических перестроек в тканях.
Сделан вывод о перспективности выбранного направления исследований, адекватности методических приемов для решения поставленных задач и о необходимости дальнейших исследований сложного баланса строго регулируемой физиологической и персистентной патологической активности кальпаинов, нарушение которого лежит в основе патофизиологии многих нейродегенеративных заболеваний.
ЗАКЛЮЧЕНИЕ
В ходе проведенного эксперимента по моделированию БА у крыс путем интрацеребрального введения пептида Аβ1-40 было изучено участие белков кальпаин / кальпастатиновой протеолитической системы в развитии нейропатологии. Об их селективной регуляции в данных условиях судили по уровню протеолитической активности основных форм кальпаинов – μ- и m-кальпаинов. Указанные кальпаины синтезируются во всех тканях и количественно превосходят другие формы фермента (в ЦНС – кальпаины 3, 5, 10) на порядки (Wuet al., 2007). μ-Кальпаин invitro активируется ионами Ca2+при микромолярных, а m-кальпаин – при миллимолярных концентрациях (Mellgen, 1980).
В мозговой ткани крыс более высокая активность кальпаинов была зарегистрирована в цитозольной фракции (свыше 90% от общей; рис. 1).
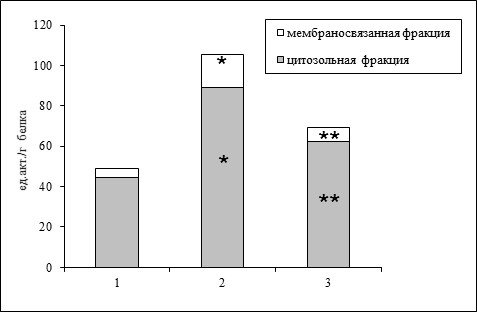
Рисунок 1. Удельная активность кальпаинов в коре больших полушарий крыс.
Здесь и на рис. 2 обозначены группы животных: 1 – контроль (ложно-оперированные); 2 – введение пептида Аβ1-40; 3 – сочетанное введение Аβ1-40 и 17β-эстрадиола.
Вместе с тем, учитывая особую роль мембранных фосфолипидов для активации этих протеиназ (Gollet al., 2003), наиболее важным показателем представляется их активность, ассоциированная с мембранными фракциями (грубой митохондриальной, микросомной, миелиновой), которая составила 9% от общей (рис. 1). Такое распределение пула кальпаинов между растворимой и мембраносвязанной фракциями клетки в разной степени сходно для большинства тканей млекопитающих (Gollet al., 2003; Kolchinskaya, Malysheva, 2004). Мы обнаружили, что в присутствии амилоидогенного пептида кальпаиновая система в коре больших полушарий активируется почти в 2 раза (рис. 1). При этом наблюдается увеличение активности мембраносвязанной фракции кальпаинов до 16% от общего пула. В наших исследованиях (Рендаков и др., 2014) было показано, что уровень общей активности кальпаинов коррелирует с интенсивностью гибели клеток нервной ткани у крыс экспериментальных групп.
Поскольку активность кальпаинов в ткани зависит от интенсивности аутокаталитической реакции(Goll etal.,2003) , с помощью метода казеиновой зимографии было качественно оценено соотношение полноразмерных и аутолизированных μ- и m-кальпаинов. Выраженная активация кальпаинов, особенно m-кальпаина (на зимограмме – белковая полоса с молекулярной массой 120 кДа), и их аутокаталитических фрагментов с молекулярной массой 118 кДа (рис. 2) у животных с экспериментальной БА является наиболее достоверным свидетельством активации кальпаиновой системы invivo. Характерно, что в целом невысокая активность μ-кальпаина (123 кДа) в нервной ткани, составляющая в норме менее 10% от уровня активности m-кальпаина,почти полностью утрачивается у крыс с экспериментальной нейродегенерацией (рис. 2), что указывает на селективную регуляцию разных форм кальпаина при развитии патологии.

Рисунок 2. Зимограмма с казеином, демонстрирующая активные фракции кальпаинов нервной ткани крыс.
Полоса с молекулярной массой 123 кДа соответствует μ-кальпаину, 120 кДа – m-кальпаину, 118 кДа – зрелым μ- и m-кальпаинам, образовавшимся путем аутолиза. Обозначения номеров дорожек см. в подписи к рис. 1.
Большинство нейропатологий, включая нейродегенерацию, ишемию, травмы мозга, а также нормальное старение, сопряжено с нарушением динамического равновесия внутриклеточного Са2+ (Bezprozvanny, 2009). Обычно общее содержание свободного Са2+ в цитоплазме поддерживается в диапазоне от 100 нМ до 1 мкМ (в покое и при стимуляции, соответственно). При БА концентрация внутриклеточного Са2+ достигает величины сотен мкМ, а локально, например в области Са2+-каналов, еще на порядок выше, что достаточно для персистентной активации не только μ-, но и m-кальпаина, которую мы наблюдаем на зимограмме. Обнаруженная нами избирательная активация m-кальпаина, которому требуется нефизиологично высокая концентрация Са2+ для активации и аутолиза, по всей видимости, объясняется избытком кальция в цитоплазме и отражает “патологическую” активацию кальпаиновой системы, аналогичную той, что наблюдается при дегенеративных процессах и в других тканях (кардиомиопатии, макулодистрофии, кахексии, миодистрофиях, ототоксичности) (Gollet al.,2003; Немова и др.,2010).
Помимо чувствительности к Са2+, изучаемые ферменты различаются субклеточной локализацией: m-кальпаин дисперсно растворен в цитозоле и ассоциирован с мембранами эндоплазматического ретикулума, а μ-кальпаин преимущественно локализован на поверхности везикул аппарата Гольджи и в небольших количествах обнаруживается в митохондриях(Gollet al., 2003; Немова и др., 2010; Hoodet al., 2010). Вероятно, это и определяет отмеченные нами различия в отклике ферментов на приток Са2+, который, как теперь известно, оказывает специфичные эффекты в зависимости от источника поступления. Концепция избирательности источников дополнительного Са2+ для индукции клеточной гибели была выдвинута Майклом Тымянски (Tymianskiet al.,1993); позже было показано, что она справедлива и для активации кальпаинов: обратный ток Са2+ через Na+/Ca2+-обменник приводит к активации кальпаинов, а приток Са2+ по другим путям, например через потенциал-зависимые Са2+-каналы, – нет (Araujoet al., 2007). Преимущественная активация m-кальпаина при изучаемом воздействии, вероятно, объясняется солокализацией ионообменника и m-кальпаина, которая увеличивает вероятность активации последнего.
Полученные результаты согласуются с рядом наблюдений. У пациентов с БА была описана аномальная активация μ-кальпаина, сконцентрированного в синаптических терминалях (Saitiet al., 1993). Избыток активной формы m-кальпаина был обнаружен в посмертных образцах префронтальной коры мозга больных деменцией альцгеймеровского типа, причем во всех специфично поражаемых болезнью зонах мозга пациентов с БА наблюдалось снижение уровня их эндогенного ингибитора, кальпастатина (Saitoet al., 1993; Nixonet al., 1994). Активация кальпаинов на фоне дефицита кальпастатина также была выявлена в мозге трансгенных мышей Tg2576, несущих мутантный вариант гена АРР человека (Vaisidet al., 2007).
Следует отметить, что у крыс, которым вводили в мозг β-амилоидный пептид, отмечалось значительное ухудшение результатов поведенческого теста (водного лабиринта Морриса). В аналогичных условиях были отмечены изменения и в других протеолитических путях, например, лизосомальной аутофагии (Рендаков и др., 2014).
СПИСОКЛИТЕРАТУРЫ
1) Бондарева Л.А., Немова Н.Н., Кяйвяряйнен Е.И. Внутриклеточная Са2+-зависимая протеолитическая система животных. М.: Наука, 2006. 304 с.
2) Немова Н.Н., Лысенко Л.А., Канцерова Н.П., Протеазы семейства кальпаинов. Структура и функции. Отногенез. 2010. Т. 41. (5): 381-389.
3) Nixon R.A. // Ageing Res. Rev. 2003. V. 2. P. 407–418.
4) Siman R., Noszek J.C. // Neuron. 1988. V. 1. P. 279-287.
5) Carragher N.O., Walker S.M., Scott Carragher L.A., Harris F., Sawyer T.K., Brunton V.G., Ozanne B.W., Frame M.C. // Oncogene. 2006. V. 25. P. 5726–5740.
6) Chakraborti S., Alam M.N., Paik D., Shaikh S., Chakraborti T. // Indian J. Biochem. Biophys. 2012. V. 49(5). P. 316-328.
7) Moldoveanu T., Gehring K., Green D.R. // Nature. 2008. V. 456. P. 404-408.
8) Averna M., De Tullio R., Capini P., Salamino F., Pontremoli S., Melloni E. // Cell. Mol. Life Sci. 2003. V. 60. P. 2669–2678.
9) Hanna R.A., Campbell R.L., Davies P.L. // Nature. 2008. V. 456. P. 409-413.
10) Berridge M.J., Bootman M.D., Roderick H.L. // Nat. Rev. Mol. Cell. Biol. 2003. V. 4(7). P. 517-529.
11) Bevers M.B., Neumar R.W. // J. Cereb. Blood Flow Metab. 2008. V. 28(4). P. 655-673.
12) АлтаеваЭ.Г., ЛысенкоЛ.А., КанцероваН.П., НемоваН.Н., ШенкманБ.С. // Докл. АН. 2010. Т. 433. № 1. С. 138-141.
13) Atherton J., Kurbatskaya K., Bondulich M., Croft C.L., Garwood C.J., Chhabra R., Wray S., Jeromin A., Hanger D.P., Noble W. // Aging Cell. 2014. V. 13. P. 49–59.
14) Abele K., Yang J. // Acta Physiologica Sinica. 2012. V. 64(5). P. 504–514.
15) Carrell R.W., Lomas D.A. Conformation disease// Lancet. 1997. V. 350. P. 134-138.
16) Hardy J. // J. Neurochem. 2009. V. 110. P. 1129-1134.
17) Grundke-Iqbal I., Iqbal K., Tung Y.C., Quinlan M., Wisniewski H.M., Binder L.I. // Proc. Natl Acad. Sci. USA. 1986. V. 83. P. 4913-4917.
18) La Ferla F. // Nat. Rev. Neurosci. 2002. V. 3. P. 862-872
19) Tydlacka S., Wang C.E., Wang X., Li S., Li X.J. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons // J. Neurosci. 2008. V. 28. P. 13285-13295.
20) Danysz W., Parsons C.G. // Brit. J. Pharmacol. 2012. V. 167. P. 324–352.
21) Berry J.N., Sharrett-Field L., Butler T.R., Prendergast M.A. // Neurosci. 2012. V. 222. P. 147–158.
22) Vaisid T., Kosower N.S., Elkind E., Barnoy S. // J. Neurosci. Res. 2008. V. 86. P. 2314-2325.
23) Vosler P.S., Brennan C.S., Chen J. // Mol. Neurobiol. 2008. V. 38. P. 78-100.
24) Bezprozvanny I. Calcium signaling and neurodegenerative diseases // Trends Mol. Med. 2009. V. 15. P. 89-100.
25) Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding // Anal. Biochem. 1976. V. 72. P. 248-254.
26) Enns D.L., Belcastro A.N. Early activation and redistribution of calpain activity in skeletal muscle during hindlimb unweighting and reweighting // Can. J. Physiol. Pharmacol. 2006. V. 84. P. 601-609.
27) Figueiredo-Petera M.E., Efthimiopoulos S., Tezapsidis N. Distinct secretases, a cysteine protease and serine protease, generate the C-termini of amyloid β-proteinase Aβ 1-40 and Aβ 1-42, respectively // J. Neurochem. 1999. Vol. 72. P. 1417-1422.
28) Goll D.E., Thompson V.F., Li H., Wei W., Cong J. Calpain system // Physiol. Rev. 2003. Vol. 83,N 3. P. 731-801
29) Grynspan F., Griffin W.R., Catalado A. Active site-directed antibodies identify calpain II as early-appearing and pervasive component of neurofibrillary pathology in Alzheimer’s disease // Brain Res. 1997. Vol. 763. P. 145-158.
30) Guttmann R.P., Johnson G.V.W. Calpain-mediated proteolysis of neuronal structural proteins // Calpain–Pharmacology and Toxicology of Calcium-dependent Protease, Taylor & Francis, Philadelphia, PA. – 1999. – С. 229-249.
31) Han P., Dou F. et al. Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: a novel excitoprotective mechanism involving modulation of tau phosphorylation //J. Neurosci. 2005. Vol. 25, N 50. P. 11542-11552
32) Hood J.L., Brooks W.H., Roszman T.L. Differential compartmentalization of the calpain/calpastatin network with the endoplasmic reticulum and Golgi apparatus // J. Biol. Chem. 2004 V. 279. P. 43126-43135
33) Kolchinskaya L.I., Malysheva M.K. Activity of calpain in subcellular fractions of the rat brain // Neurophysiol. 2004. V. 36. P. 265-271.
34) Kusakawa G.-I., Saito T., Oonuki R. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase activator to p25 // J. Biol. Chem. 2000. Vol. 275. p. 17166-17172.
35) Lee M.-S., Kwon Y. T., Li M., Peng J., Friedlander R.M., Tsai L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain // Nature. 2000. Vol. 405. P. 360-364.
36) Leissring M.A., Akbari Y., Fanger C. M. et al. Capacitative calcium entry deficits and elevated luminal calcium in mutant presenilin-1 knockin mice // J. Cell. Biol. 2000. Vol. 149. P. 793-797.
37) Litersky J.M., Johnson G.V. Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain // J. Biol. Chem. 1992. Vol. 267. P. 1563-1568
38) Marcilhac A. Intracellular signaling pathways, apoptosis and neurodegenerative diseases // Psychologie & neuropsychiatrie du vieillissement. 2004. Т. 2. №. 3. С. 203-214.
39) Marcum J. L., Mathenia J. K., Chen R., Cuttmann R.P. Oxidation of thiol-proteases in the hippocampus of Alzheimer’s disease // Biochem. Biophys. Res. Commun. 2005. Vol. 334, N 2. P. 342-348
40) Mellgren R.L. Canine cardiac calcium-dependent proteases: resolution of two forms with different requirements for calcium // FEBS Lett. 1980. V. 109. P. 129-133.
41) Morris R.G.M. Spatial localization does not require the presence of local cues // Learn. Motiv. 1981. V. 2. P. 239-260.
42) Nixon R.A., Mohan P. Calpains in the pathogenesis of Alzheimer’s disease // Calpain: Pharmacology and toxicology of calcium-dependent protease / Ed. by K.K.W. Wang, P.-W. Yuen. Philadelphia (PA): Taylor and Francis, 1999. P. 229-249.
43) Nixon R.A., Saito K.I., Grynspan F., Griffin W.R., Katayama S., Honda T., Mohan P.S., Shea T.B., Beermann M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease // Ann. N-Y Acad. Sci. 1994. V.747. P. 77-91.
44) Rawlings N.D., Barrett A.J., Bateman A. MEROPS: the peptidase database // Nucleic Acids Res. 2012. V. 40. P. D343-D350.
45) Saito K-I., Elce J.S., Hamos J.E., Nixon R.A. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer’s disease: a potential molecular basis for neuronal degeneration // Proc. Nat. Acad. Sci. USA. 1993. Vol. 90. P. 2628-2632.
46) Selkoe D.J. Alzheimer disease: genes, proteins, and therapy // Physiol. Rev. 81. 2001. Vol. 81. P. 741-766.
47) Tymianski M., Charlton M.P., Carlen P.L., Tator C.H. Source speci1city of early calcium neurotoxicity in cultured embryonic spinal neurons // J. Neurosci. 1993. V. 13.P. 2085-2104.
48) Vaisid T., Kosower N.S., Katzav A., Chapman J., Barnoy S. Amyloid b peptide toxicity in differentiated PC12 cells: Calpain-calpastatin, caspase, and membrane damage // Neurochem. Int. 2007. V. 51. P. 391-397.
49) Wu H.Y., Tomizawa K., Matsui H. Calpain–calcineurin signaling in the pathogenesis of calcium-dependent disorder // Acta Med. Okayama. 2007. V. 61. P. 123-137.
50) Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved “calpain-cathepsin cascade” from nematodes to primate // Cell Calcium. 2004. Vol. 36, N 3-4. P. 285-293.
51) Коросов А.В., Горбач В.В. Компьютерная обработка биологических данных. Петрозаводск: ПетрГУ, 2007.
52) Немова Н.Н., Лысенко Л.А., Канцерова Н.П. Протеиназы семейства кальпаинов. Структура и функции // Онтогенез. 2010. Т. 41. С. 381-389.
53) Рендаков Н.Л., Лысенко Л.А., Люпина Ю.В., Шарова Н.П., Сельверова Н.Б., Немова Н.Н. Роль лизосомальных протеиназ и эстрадиола в нейродегенерации, индуцированной бета-амилоидом // Докл. АН. 2014
0 комментариев