Содержание
Введение. ………………………………………………………………………...2
1. История возникновения GMP. ……………………………………………..4
1.1 Предпосылки появления GMP. ………………………………………..4
1.2 Первые правила GMP. …………………………………………………5
1.3 История GMP в России. ………………………………………………...7
2. Значение Правил GMP. ……………………………………………………..9
3. Стандарты, относящиеся к Правилам GMP. …………………………….13
4. Некоторые принципы Правил GMP. ………………………………………17
4.1 Принципы GMP для активных фармацевтических субстанций. …...17
4.2 Принципы GMP для вспомогательных веществ……... ………………18
5. Фармацевтические предприятия, работающие по Правилам GM в РФ...19 6. Стратегия "Фарма 2020"……………………………………………………25
6.1 Цели, задачи и приоритеты государственной политики РФ по развитию национальной фармацевтической промышленности…………….25
6.2 Ожидаемые результаты реализации Стратегии……………………..27
6.3 Проблемы фармацевтической отрасли……………………………… 29
6.4 Основные мероприятия и ожидаемые результаты реализации
Стратегии…………………………………………………………………33
6.5 Объем и источники финансирования мероприятий Стратегии……..41
7.Исследование доли выпускаемой продукции отечественных производителей в ассортименте товаров аптек базы практики……………..43
7.1.Анализ доли препаратов ЗАО "Эвалар"…..………...……………….43
7.2 Анализ доли препаратов ОАО "Нижфарм" ………………………..48
7.3 Анализ доли препаратов ОАО ХФК "Акрихин"…..………………..52
8.Заключение……………………………………………………………………56
9.Список использованных источников………………………………………..59
Введение
Разработка и производство качественных лекарственных препаратов, а также изделий медицинского назначения является приоритетным направлением мировой фармацевтической науки и практики. Для поддержания качества продукции на этапе производства у многих производителей используются различные меры и способы, которые позволяют контролировать на стадиях и операциях продукцию путем отбора образцов с конкретной партии. Учитывая особенности производства лекарственных препаратов, проконтролировать полностью всю продукцию, которая находятся в конкретной партии, не разрушая при этом саму лекарственную форму технически невозможно, т.к. вскрытый флакон, разрушенная капсула или таблетка уже не могут использоваться потребителем. Поэтому часто контроль проводится на ограниченной выборке лекарственных форм из конкретной серии продукции.
Очевидно, что использование такого похода не может в полной мере гарантировать качество продукции, поэтому учеными были разработаны правила надлежащей производственной практики (Good manufacturing practice – GMP) которые содержат свод необходимых минимальных требований к организации производства фармацевтической продукции и изделий медицинского назначения, направленные на обеспечения их качества. В связи с этим само понятие «качество продукции» характеризуется намного шире, чем просто соответствие требованиям фармакопейных статей или конкретных спецификаций. В производственном процессе должны быть исключены техническое перепутывание действующих и вспомогательных веществ, попадание в них посторонних продуктов, использование некачественных ингредиентов и материалов и др. Главной целью надлежащей производственной практики является гарантированное соответствие состава лекарственного средства, отсутствие в нем различных загрязнений, корректность маркировки, неизменность и стабильность свойств в процессе хранения в течении установленного срока годности.
Цель дипломной работы: исследование доли выпускаемой продукции отечественных производителей в ассортименте товаров аптек базы практики.
Объект исследования: товары отечественных производителей лекарственных препаратов, работающих по правилам GMP.
Предмет исследования: занимаемая доля товаров отечественных производителей, работающих по правилам GMP, в ассортименте лекарственных препаратов аптек базы практики.
В связи с целью работы были поставлены следующие задачи:
1. Дать определение термину «GMP».
2. Изучить историю правил GMP.
3. Проанализировать перечень документов, регламентирующих правила GMP.
4. Изучить некоторые основные принципы правил GMP.
5. Систематизировать сведения о фармацевтических предприятиях в России, имеющих сертификат соответствия правилам GMP.
6. Выявить проблемы, связанные с внедрением правил GMP в России.
7. Проанализировать ассортимент лекарственных препаратов в аптеках базы практики.
8. Рассчитать долю лекарственных препаратов отечественных производителей, работающих по правилам GMP, в общем ассортименте лекарственных препаратов и проанализировать спрос на них.
1. История возникновения GMP
1.1 Предпосылки появления GMP
После завершения Второй мировой войны в Японии, Западной Европе и Соединённых Штатах Америки (США) наблюдался научно-технический прорыв во всех отраслях, в том числе и фармации. Необходимо отметить, что наиболее успешными в этом прорыве были США, т.к. эта страна наименее пострадала от последствий военных действий, а развитие других стран было несколько замедлено в связи с необходимостью восстановления промышленности после военных разрушений. [1]
В это время на больших промышленных химических предприятиях разрабатывались новые активнодействующие ингредиенты для лекарственных форм с различным терапевтическим действием, например, таким как антимикробное, противовирусное, сердечно-сосудистое, психотропное и др. Это способствовало расширению ассортимента лекарственных форм. Другие предприятия, не имея всех ресурсных возможностей использовали в качестве действующего вещества известные субстанции, но для того чтобы запатентовать производство препарата, производитель незначительно химически модифицировал формулу активнодействующего вещества. Это позволяло акцентировать в рекламе новизну формулы препарата и предполагать, что этот препарат лучше в сравнении с оригинальным. [4]
На больших химических предприятиях, которые производили гербициды, красители, удобрения, а также фармацевтические субстанции, не всегда должным образом контролировалось качество фармацевтических субстанций. В основном это касалось санитарного состояния, риска дополнительной контаминации фармацевтических субстанций другими субстанциями, производимые этим заводом.
На предприятиях меньшего масштаба, где было сосредоточено только фармацевтическое производство, провизоры и фармацевты придерживались культуры так называемого «фармацевтического порядка», но по сравнению с большими концернами химических заводов эти предприятия могли отставать в оснащении лабораторий по контролю качества. [5]
В середине прошлого столетия начали появляться промышленные предприятия, которые не разрабатывали новые препараты или модифицировали молекулы известных препаратов, а повторяли или копировали известные препараты и выпускали их на рынок только под измененными названиями. Их называли «препараты-дженерики». Эти производители зачастую не располагали опытом фармацевтического производства или ресурсами и возможностями больших концернов. Основным инструментом конкуренции на фармацевтическом рынке для них была цена и тотальная экономия на всем, в том числе и на качестве. На рынке стали появляться препараты сомнительного качества, у которых могла быть пересортица этикеток или случайное загрязнение активной субстанции другими субстанциями. Все вышеперечисленное способствовало развитию новой системы качества лекарственных препаратов, которая позволяла бы комплексно обеспечивать качество готовой продукции включая профилактику механических ошибок и отклонений в процессе производства. [3]
1.2 Первые правила GMP
Первые правила надлежащей производственной практики были созданы в Соединённых Штатах Америки в 1963 году и представляли собой всего лишь двухстраничный документ, суть которого сводилась к тому, что производители придерживались требований «фармацевтического порядка». Главным образом это было направлено на производителей – аутсайдеров отрасли, в основном это были предприятия, которые производили «препараты-дженерики». [10]
Случай с препаратом «Талитомид» только ускорил процесс перехода предприятий к новым нормам контроля качества. Суть была в следующем, препарат «Талитомид» позиционировался на рынке Европы как снотворное и уменьшающее тошноту во время беременности. Выпуская на рынок данный препарат, никто не знал, что у него кроме выраженного терапевтического действия существуют значительные побочные эффекты. Он обладал тератогенным эффектом, вызывая уродство плода. Рожденные дети были с деформированными конечностями. Количество полученных врождённых уродств, связанных с приемом данного препарата насчитывало свыше десяти тысяч. На рынок США данный препарат так и не попал благодаря ученой Фрэнсис Келси, которая приняла решение о запрете использования данного препарата. В результате на законодательном уровне были приняты нормы, которые обязывали производителей предварительно проводить клинические испытания препаратов на животных перед использованием их на людях. Производители должны были обязательно предупреждать пациентов, принимающих участие в исследовании, получать письменное согласие перед употреблением препарата.
Таким образом, был создан механизм, который подтверждал эффективность препарата перед выходом его на рынок. Производители в этой схеме должны в обязательном порядке сообщать о побочных эффектах препаратов. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (Food & Drug Administration - FDA) наделили полномочиями по регуляции рекламы рецептурных препаратов. [2]
Необходимо отметить, что первоначально другими странами не предпринимались меры по распространению первых правил GMP, разработанных в США в области нормирования производства фармацевтических препаратов. После того, как правила GMP были признаны Всемирной организации здравоохранения (ВОЗ), их начали активно использовать и в других государствах.
Первоначально правила GMP разрабатывались в каждой стране отдельно, большего успеха в этом достигли США и Великобритания. Со временем было принято решение о консолидации правил под единый стандарт, который бы мог использоваться на межгосударственном уровне. Это было узаконено в Европе в 1970 г., путем заключения Конвенции по инспектированию фармацевтических предприятий (Pharmaceutical Inspection Convention - PIC). А в 1989 году были приняты правила GMP Европейского Союза. В 2010 г. Китай принял правила GMP, основанные на GMP EC и ВОЗ, но с добавлением аспектов по традиционной китайской медицине.
1.3 История GMP в России
Развитие GMP в России было обусловлено экономикой. До 1991 года химико-фармацевтические предприятия, как и остальные предприятия промышленности получали план от государства на выполнение. Причем производители не заботились о рынке товаров, выявляя его потребности, для них главной целью было выполнить план. Государство аккумулировало в едином лице роль заказчика, поставщика, производителя и контролера. Функционирование данной системы обеспечивалось множественными бюрократическими организациями, которые прежде всего ориентировались на внутренний рынок. Внедрение новых технологий, международных норм и правил требовало финансовых затрат, а если учесть, что это были предприятия не оборонной промышленности, финансирования не всегда хватало. Стандарты «подгонялись» под возможности производств с трактовкой так называемой «национальной специфики». [2]
В начале девяностых годов ситуация изменилась, в связи с изменением функций государства и переходом на рыночную экономику. Производители, попав в новые условия рынка, начинали постепенно адаптироваться к новым условиям. На уровне государства были дискуссии: внедрить правила GMP EC или же использовать нормы с «национальной спецификой». В результате через некоторое время вступил в силу Федеральный закон «О техническом регулировании» которым был установлен приоритет международных норм. Далее уже в 2004 году был принят ГОСТ Р 52249-2004 «Правила производства и контроля качества лекарственных средств» который был идентичен GMP EC. Затем был принят ГОСТ Р 52249-2009, который являлся идентичным переводом правил GMP EC от 2009 года. [1]
2. Значение правил GMP
Правила GMP - это комплексная система требований к организации производства и контроля качества фармацевтических средств.
Правила GMP можно объединить в несколько групп:
1. В сфере производства и общего управления компанией.
1.1 До начала производства обеспечиваются все необходимые для этого условия, включая:
• квалифицированный персонал;
• достаточные помещения;
• соответствующее оборудование;
• надлежащие исходные материалы;
• нормативную документацию и протоколы анализа;
• нормы складирования и транспортировки;
• установленные порядки контроля качества.
1.2 Все производственные процессы валидированы, то есть проверены на надежность, утверждены, обоснованы и зарегламентированы.
1.3 Все производственные операции подвергаются строгому протоколированию в соответствии с установленными порядками на каждом отдельном предприятии.
1.4 Отгрузка и хранение товара строго протоколируются, с возможностью проследить ход каждой серии каждого препарата до возможности отзыва любой серии из сбыта.
1.5 Рассмотрение всех жалоб в отношении проданного товара.
2. В сфере контроля качества.
2.1 Отдел контроля качества не зависим от производства, обеспечен собственными ресурсами для проведения качественных анализов.
2.2 Каждый контрольный этап: отбор проб, анализ и испытания проводятся уполномоченными сотрудниками в соответствии с утверждёнными должностными инструкциями и спецификациями.
2.3 Выпуск готовой продукции может быть выпущен в реализацию только после утверждения досье, аналитического листа или другого подтверждающего документа.
2.4 Сохраняется достаточное для повторного контроля количество образцов сырья, исходных материалов и готовой продукции.
2.5 Предприятие регулярно подвергается самоинспектированию.
Контроль производственного процесса соответствующим образом документированный, обеспечивает воспроизводство лекарств, однородных по свойствам как внутри серий, так и между ними, и, следовательно,
гарантирует стандартность фармацевтических качеств и безопасность препарата. Конечный продукт должен быть испытан в Отделе технического контроля (ОТК) на соответствие физико-химических и биологических свойств требованиям Государственной Фармакопеи или другого утвержденного стандарта нормативно-технической документации (НТД). [8]
Наиболее общими для всех GMP являются разделы, касающиеся производства стерильных лекарственных форм. Они содержат следующие требования:
1. В чистой и асептической зонах число рабочих не должно превышать потребностей одной смены.
2. Допуск к работе в чистой зоне имеют только рабочие, прошедшие предварительную подготовку.
3. Одежда рабочих должна иметь метки допуска работы в чистых зонах (нашивки, цвет и др.). Одежда должна соответствовать назначению чистой и асептической зоны и включать в себя обувь, головной убор, перчатки и комплект брюки - халат.
4. Интервал между началом приготовления раствора и его стерилизацией должен составлять не более 3 часов.
5. Активность в чистой и асептической зонах должна быть сведена до минимума, чтобы избежать перемещения частиц и микроорганизмов.
6. Для каждого цикла стерилизации должны контролироваться следующие данные: время, давление, температура и влажность. Каждый цикл стерилизации контролируется соответствующими биологическими или химическими индикаторами.
7. В число проб, взятых для определения стерильности, обязательно должны входить пробы, взятые в начале и в конце работы, после значительного перерыва в работе, а также из потенциально наиболее холодной части загрузки.
8. Между помещениями различных классов чистоты должна поддерживаться постоянная разность давления в 3-5 мм вод. ст. При этом в помещениях более высоких классов чистоты давление должно быть выше.
9. Наиболее ответственные технологические операции должны быть защищены установками ламинарного потока стерильного воздуха.
10. Все открытые поверхности (стены, потолок, пол) должны быть гладкими, легко моющимися и устойчивыми к действию дезинфицирующих средств.
11. Между помещениями различных классов чистоты должны быть переговорные устройства.
12. Помещения для стерилизации должны быть спроектированы и построены таким образом, чтобы не допускалась возможность смешивания простерилизованных и непростерилизованных материалов и лекарственных веществ.
13. Оборудование должно быть сконструировано и размещено таким образом, чтобы обеспечить его подготовку к работе, эксплуатацию и обслуживание.
14. Материалы оборудования не должны реагировать с используемыми сырьем и материалами.
15. Оборудование и инструменты должны регулярно подвергаться мойке, обработке дезинфицирующими средствами и (или) стерилизации, а также профилактическим осмотрам.
16. В производстве парентеральных лекарственных средств следует избегать применения фильтров, выделяющих волокна.
17. Весь персонал должен периодически проходить переподготовку и медицинские осмотры.
18. Хранение воды, используемой для изготовления парентеральных растворов большого объема, осуществляется при постоянной циркуляции при 85°С. Если хранение осуществляется при температуре окружающей среды, срок хранения воды - 24 часа, после чего она сливается. Контроль воды на пирогенность проводится 1 раз в неделю. [11]
Правила GMP России регламентируют, какие требования должны быть соблюдены в процессе промышленного производства лекарственных препаратов. Каким же образом они должны быть реализованы на практике, решает производитель.
3. Стандарты, относящиеся к Правилам GMP
Под стандартом GMP (good manufactured practice) понимают систему норм и указаний, направленных на надлежащее производство лекарственных средств, медицинских изделий, пищевых добавок и витаминов. Стандарт GMP отражает комплексный подход в оценке производства и лабораторной проверки лекарственных средств, в отличие от общепринятой практики выборочной проверки продукта производства.
В целом, стандарт GMP состоит из большого числа показателей, требующих соответствия для выпуска той или иной продукции, начиная от материала, из которого сделаны стены в цеху и заканчивая стандартными протоколами мытья рук для работников чистой зоны. Нормы GMP для фармацевтических предприятий устанавливают параметры для каждого производственного этапа - от материала, из которого сделаны стены в цеху и заканчивая стандартной операционной процедурой обработки рук в нестерильном помещении. Если обобщить, то GMP - сумма строительных, пожарных, санитарных норм и ГОСТов, обеспечивающих производство качественных лекарственных средств, отвечающих международным нормам. Однако, GMP включает в себя также требования, не предусмотренные российскими стандартами: например, работа в так называемых «чистых зонах» - помещениях с ламинарным потоком воздуха и особым режимом санитарной обработки. В СССР такие помещения строились для производства микросхем, в то время как формат GMP требует фасовать даже нестерильные лекарственные средства в таких помещениях. [4]
В настоящее время основными элементами концепции GMP являются:
• соответствие всей технологической и контрольной документации на производстве содержанию регистрационного досье на соответствующий препарат;
• жесткий контроль за соблюдением правил, предполагающий не только декларированность, но и фактическое применение санкций к предприятиям-нарушителям.
Следует отметить, что в отличие от норм ISO, которые носят рекомендательный характер, требования GMP обязательны к исполнению для защиты населения от некачественных лекарственных средств. Исполнение требований GMP гарантирует производство качественной фармацевтической продукции:
• Формат GMP предполагает комплексный подход в производстве, анализе и контроле качества исходных материалов и готовых препаратов.
• Формат GMP включает нормативные характеристики помещений, санитарной обработки, технические характеристики для помещений и оборудования, задействованных в производстве лекарственных средств.
• Нормы GMP содержат требования к инженерно-техническому обеспечению производства и контролю стерильности.
• Формат GMP требует обязательной валидации, методов исследований, записей результатов и составление отчётности.
• Также GMP предполагает составление и хранение документации на каждую изготовленную серию препаратов, как в бумажном, так и в электронном виде.
• Нормы GMP устанавливают стандарты качества к герметичности, упаковке, маркировке, хранению изготовленных лекарственных средств. [4]
Стандарты, относящиеся к исследованиям и производству:
Стандарт GEP (англ. «Good Engineering Practice», Надлежащая инженерная практика) – совокупность проверенных инженерных методов и стандартов, применяемых в течение жизни проекта в целях получения адекватных и приемлемых по цене инженерных решений.
Стандарт GMP (англ. «Good Manufacturing Practice», Надлежащая производственная практика) - совокупность Российских и международных норм и правил, определяющих параметры производства лекарственных средств, медицинских устройств, изделий диагностического назначения, продуктов питания, пищевых добавок, активных ингредиентов.
Стандарт GLP (англ. «Good Laboratory Practice», Надлежащая лабораторная практика) - система норм, правил и указаний, обеспечивающих согласованность и достоверность результатов лабораторных исследований.
Нормы GLP изначально появились в США, а затем усилиями Организации экономического сотрудничества и развития стали применяться на международном уровне.
Благодаря строгой системе ведения документации правила GLP позволяют специальным инспектирующим органам проследить и восстановить весь ход проводимых исследований.
ГОСТ Р 52249-2009 (Государственный стандарт Российской Федерации) устанавливает требования к производству и контролю качества лекарственных средств для человека и животных.
Стандарт ГОСТ Р 52249-2009 заменил действовавший ранее ГОСТ Р 52249-2004 и идентичен Правилам производства лекарственных средств для человека и животных Европейского Союза (ЕС Guideto Good Manufacturing Practicefor Medicinal Products for Human and Veterinary Use) по состоянию на 31.01.2009г. [1]
Правила организации производства и контроля качества лекарственных средств были утверждены приказом Минпромторга России 14 июня 2013 г. N 916.
Стандарт регламентирует производство всех видов лекарственных средств, устанавливает требования к контролю качества, а также специальные требования к производству отдельных видов лекарственных средств и активных фармацевтических субстанций.
Стандарты качества, относящиеся к дистрибьюции, хранению и лекарственному обеспечению населения:
Стандарт GSP (англ. «Good storage practices for pharmaceuticals», Надлежащая практика хранения фармацевтической продукции) – устанавливает комплекс мер, призванных обеспечить правильное хранение и транспортировку фармацевтической продукции.
Стандарт применяется с учетом вида деятельности предприятия и ориентирован на всех работников, имеющих отношение к хранению, транспортировке и распространению фармацевтической продукции. Стандарт дополняет собой такие руководства как GDP, GMP, Международную Фармакопею.
Стандарт GPP (англ. «Good Pharmacy Practice», Надлежащая аптечная практика) – комплекс норм и правил, призванных обеспечить надлежащее качество фармацевтических услуг, оказываемых аптечными работниками населению.
Данный стандарт предназначен для работников аптек и определяет роль фармацевта в системе здравоохранения.
Стандарт GDP (англ. «Good Distribution Practice», Надлежащая дистрибьюторская практика) устанавливает единый подход к организационному процессу оптовой реализации лекарственных средств и направлен на обеспечение качества препаратов на всем пути от производителя до розничной сети и медицинских организаций. В его основу положены принципы надлежащей дистрибьюторской практики, принятые в Европейском Сообществе и рекомендованные Всемирной Организацией Здравоохранения.
Соблюдение указанного стандарта обеспечивает: качество и безопасность лекарственных средств, медицинской техники и изделий медицинского назначения, гарантированные производителем; поступление лекарственных средств, медицинской техники и изделий медицинского назначения без изменений их свойств в розничную сеть и медицинские организации. [10]
Целью стандарта является сохранение качества товара при его движении от производителя до потребителя.
4. Некоторые принципы Правил GMP
Правила GMP основаны на следующих принципах:
· системный подход;
· профилактическая направленность;
· гибкость в способах выполнения требований;
· обязанность производителя представить доказательства адекватности избранного им метода выполнения требований (валидация).
4.1 Принципы GMP для активных фармацевтических субстанций
Общие требования к производству активных фармацевтических субстанций (АФС) даны в части II ГОСТ Р 52249-2009. Этот документ предусматривает три уровня защиты материалов и АФС в процессе производства. Эффективным средством защиты от перекрёстных загрязнений и защиты от вредных веществ являются перепады давления воздуха. В производстве стерильных АФС перепады давления служат для разделения зон с различными классами чистоты.
1. Защита от перекрёстных загрязнений. Требования к защите различаются для специализированных производств у которых есть многономенклатурные позиции. Частицы воздуха с таких помещений могут оседать на стенках воздуховодов и проходить даже через НЕРА фильтры.
2. Материал для воздуховодов. Как правило используется оцинкованная или нержавеющая сталь. Воздуховоды нельзя окрашивать.
3. Микроклимат.Следует предусматривать контроль температуры и влажности с регистрацией их изменений.
4. Требования к пару. Для увлажнения воздуха в кондиционере следует использовать чистый пар. Пар не должен содержать масел и химических соединений, от которых не защищает НЕРА фильтр.
5. Аттестация производств АФС. При аттестации следует обратить внимание на критические стадии производства АФС.
4.2 Принципы GMP для вспомогательных веществ:
1. Контроль посторонних включений и загрязнений. Несмотря на то что вспомогательные вещества производятся на химических производствах в закрытых оборудованиях, необходимо предусматривать защиту от загрязнений из окружающей среды.
2. Характеристика и свойства вспомогательных веществ. Производство вспомогательных веществ должно быть стабильным и обеспечивать неизменный состав лекарственного средства согласно спецификации. Это особенно важно для стерильных и апирогенных вспомогательных веществ.
3. Неизменность состава вспомогательных веществ от серии к серии и контроль изменений. Необходимо учитывать тот факт, что при производстве вспомогательных веществ технологический процесс может меняться. Нужно это учитывать с помощью системы изменений.
4. На каждую серию должен оформляться протокол серии. Весь процесс производства должен прослеживаться по протоколу серии.
Требования GMP для вспомогательных веществ аналогичны требованиям GMP к производству активных фармацевтических субстанций и готовых лекарственных средств и распространяются на:
- документацию и контроль изменений;
- ответственность и планирование;
- персонал, его подготовку и гигиену;
- здание, помещение и оборудование;
- технологические материалы (воду, пар, газы), компьютерные системы и инфраструктуру;
- защиту от насекомых, грызунов, птиц;
- уборку, контроль параметров окружающей среды;
- производство и склады;
- реализацию продукции;
- анализ недостатков и работу по их устранению;
- проведение аудита с указанием контрольных точек. [7]
5. Фармацевтические предприятия, работающие по Правилам GMP
Большинство российских предприятий отстает с внедрением правил GMP на несколько десятилетий от уровня индустриальных стран. В качестве причины запаздывания официально признается только одна – недостаток финансовых средств. Между тем, как представляется, немалую роль играет и психологическая неготовность ведущих специалистов отрасли к переменам.
Российская фармацевтическая промышленность должна перейти на стандарты GMP до 31 декабря 2013 года. С 1 января 2014 года лицензии на производство ЛС выдаваются только при условии соответствия правилам GMP, а предприятия, уже получившие бессрочные лицензии, должны их подтвердить, пройдя государственную инспекцию. Этот срок был установлен еще в 2010 году Федеральным законом № 61 «Об обращении ЛС», и предполагал, что в течение трех лет будут приняты все необходимые нормативно-правовые акты, обеспечивающие условия и процедуру подтверждения производства правилам GMP, проведены проверки и выданы заключения соответствия. [5]
Затем Минпромторг внес предложение о включении в «ФЗ» № 61 «Об обращении лекарственных средств» таких понятий как «сертификат GMP» и «надлежащая производственная практика».
Планируется, что проверка всех фармпредприятий специалистами Минпромторга будет завершена к 2018 году. Импортеры лекарств должны будут пройти проверку на соответствие стандартам GMP к 2020 году. Принципиальное решение о проверках принято и зафиксировано в готовящихся сейчас поправках к Закону «Об обращении лекарственных средств» ко второму чтению. Пока еще точные сроки проведения проверок иностранных фармпроизводителей обсуждаются. Также прорабатывается вопрос о взаимном признании результатов проверок — оно может быть достигнуто только соглашениями между странами. [3]
Некоторые фармацевтические предприятия, у которых есть сертификат GMP:
1. Компания ОАО «Нижфарм» (STADA CIS), г. Нижний Новгород, получила Российский сертификат №1 GMP (Good Manufacturing Practice), разработанный и утвержденный Минпромторгом России. В конце 90-х годов ОАО «Нижфарм» после привлечения инвестиций Европейского банка реконструкции и развития построила «с нуля» цех по производству таблетированных лекарственных форм в соответствии с GMP.
2. ЗАО «Эвалар», Алтайский край, г. Бийск. По результатам аудита, проведенного NSF, получен сертификат на соответствие требованиям GMP.
3. ОАО «Химико-фармацевтический комбинат “Акрихин”» - один из крупнейших производителей готовых лекарственных средств в России. С самого основания предприятие активно развивалось, накапливая опыт в производстве ГЛС. Сегодня оно имеет репутацию надежного партнера и производителя продукции высокого качества.
В настоящее время комбинат выпускает около 170 наименований наиболее востребованных препаратов 24 основных фармакотерапевтических групп. Около 40% препаратов включено в Перечень жизненно необходимых и важнейших лекарственных средств Минздрава РФ.
«Акрихин» стал первым отечественным предприятием, начавшим внедрение международных стандартов GMP в 1997 году. В 2002 году производственные линии сердечно-сосудистых препаратов и мазей получили Свидетельство № 1 о соответствии производства стандартам отрасли ОСТ 42-510-98 (GMP), выданное Минпромнауки и технологий РФ.
4. Фармацевтическая компания «ОЗОН» один из лидеров отечественного фармацевтического рынка по производству дженериков - аналогов патентованных готовых лекарственных средств (ГЛС). Стандарты GMP (Good Manufacturing Practice) позволяют компании производить препараты высокого качества, доступные широким слоям населения. Сегодня объем производства компании ОЗОН составляет более 150 млн. упаковок в год, а продуктовый портфель включает в себя более 60 наименований ГЛС. С каждым годом число выпускаемых препаратов растет, увеличиваются объемы производства. Неизменными остаются высокое качество и доступность выпускаемой продукции. Сотрудники, занятые на производстве, проходят обучение для соответствия требованиям GMP.
5. На предприятии ОАО «Фармстандарт-Уфавита» начато строительство цеха по производству цитостатических препаратов в соответствии с европейскими стандартами GMP (Good Manufacturing Practice – «Надлежащая производственная практика»). Компания планирует начать производство современных цитостатических препаратов как собственного производства, так и выпускаемых в рамках содружества с иностранными фармацевтическими компаниями.
6. ЗАО «ФармФирма «Сотекс» - современный производитель лекарственных средств, работающий в соответствии с требованиями GMP EU. Завод компании, располагающийся в Сергиево-Посадском районе Московской области, является одним из наиболее высокотехнологичных и инновационных фармацевтических предприятий в России. Компания «Сотекс» имеет производственное предприятие, изначально построенное в соответствии с требованиями GMP и запущенное в эксплуатацию в 2005 г. На первом этапе работы предприятия технологические процессы GMP «обкатывались» на производстве дженериков. В дальнейшем компания получила возможность привлечь контракты на производство по лицензии препаратов ряда иностранных компаний, в частности Nycomed, Sanofi-Aventis, Pierre Fabre, Bayer, Lek (сейчас Sandoz, входит в структуру компании Novartis), Croma Pharma. Перенос производства препаратов указанных компаний на площадку «Сотекса», а также ежегодные аудиты качества со стороны партнеров по лицензионному производству подтверждают высокие стандарты производственных процессов российского фармпредприятия. В 2007 году «Сотекс» запустил производство собственных брендов.
7. По пути строительства фармпредприятия, изначально соответствующего стандартам GMP, пошла также Компания «ЗиО-Здоровье» (ГК Actavis), уже в 2006 г. получившая сертификат, подтверждающий полное соответствие условий производства правилам GMP EU.
8. Тот же путь избрала Компания «Канонфарма продакшн», имеющая высокотехнологичное производство полного цикла, спроектированное, построенное и оснащенное в соответствии с требованиями GMP.
В настоящее время целесообразно создавать новые предприятия, оснащенные современным оборудованием, при наличии “гибких технологических схем” с размещением на них производства современных, высокоэффективных лекарственных препаратов (т. е. весь производственный цикл — от субстанции до готовых лекарственных препаратов). При этом выпускаемые препараты должны относиться не только к жизненно необходимым и важнейшим, но и к препаратам, закупаемым за рубежом на значительные суммы. Потенциальным источником для отечественных предприятий могут стать иностранные инвестиции. Западные компании могли бы инвестировать значительные средства в российскую фармацевтическую промышленность и науку. Однако иностранные инвесторы пока занимают выжидательную позицию.
Получение сертификата GMP в России это трудоемкий процесс, т.к. отсутствует четко выстроенная действующая государственная система нормативно-правового регулирования, соответствующая существующим международным требованиям.
Осуществлять выдачу заключений о соответствии производителей лексредств требованиям GMP поручено Министерству промышленности и торговли России. Но при этом ведомство не уполномочено разрабатывать и утверждать порядок проведения проверок и выдачи заключений о соответствии правилам. Российским производителям не известны ни график проверок, ни официальные тарифы на стоимость услуг по инспектированию.
Процедура подтверждения соответствия требованиям GMP включает такие этапы как:
1. Подача заявления на выдачу сертификата.
2. Проверка и обработка поданного заявления.
3. Инспектирование производства лекарственных средств.
4. Принятие решения о выдачи документа в соответствие с требованиями GMP.
5. Внесение изменений в действующий документ о соответствии производства требованиям GMP.
Не маловажным является то, что сертификация GMP является дорогостоящим процессом, что может привести к появлению двойных стандартов. Так как инструкция, регламентирующая несуществующий процесс или нереальные условия его выполнения, провоцирует ведение ложных записей протоколирования. Очевидно, что только при полном соответствии требований инструкций реальным возможностям процесса и, соответственно, наоборот система качества может быть внедрена и функционально жизнеспособна.
Сохраняются отдельные недостатки в отраслевом законодательстве. В конце 2013 года были приняты поправки в Федеральный закон "Об обращении лекарственных средств" (61-ФЗ). Были внесены существенные изменения в процедуры выдачи разрешений на проведение клинических исследований и регистрации новых препаратов, которые еще предстоит проанализировать. Однако поправки мало затронули практику регулирования фармацевтического производства в форме лицензирования и инспектирования площадок на соответствие правилам GMP. Обновленным законом предусмотрен лицензионный контроль в сфере производства лекарственных средств.
Одной из основных проблем является отсутствие правовых основ для создания инспектората по GMP, отвечающего всем международным требованиям. Такая структура должна иметь собственную систему качества, утвержденные надлежащим образом положение об инспекторате, должностные инструкции для инспекторов, стандартные операционные процедуры по проведению обследований и оформлению их результатов. Пока нет правовой основы для создания инспектората по GMP, отвечающего международным требованиям.
6.Стратегия «Фарма 2020»
6. Стратегия «Фарма 2020»
«Фарма—2020» (Стратегия развития фармацевтической промышленности Российской Федерации на период до 2020 г.) разработана Министерством промышленности и торговли РФ в 2008 г. Стратегия призвана определять пути реализации приоритетных направлений развития фармпроизводства России, быть основой для государственно-частного партнерства по вопросам развития фармацевтической промышленности, обеспечивать согласованность действий органов государственной власти по направлениям развития отрасли, определять векторы разработки и корректировки нормативно-правовой базы фармацевтической промышленности и служить основой для принятия государственных решений по разработке и реализации целевых программ и проектов развития отрасли.
6.1. Цели, задачи и приоритеты государственной политики Российской Федерации по развитию национальной фармацевтической промышленности
Основной целью государственной политики Российской Федерации по развитию национальной фармацевтической промышленности на период до 2020 года является создание условий для ее перехода на инновационную модель развития, что должно привести к росту обеспеченности населения, учреждений здравоохранения и Вооруженных Сил Российской Федерации,
федеральных органов исполнительной власти, в которых законом предусмотрена военная и приравненная к ней служба, лекарственными средствами отечественного производства, при общем увеличении обеспеченности нуждающихся лекарствами до среднеевропейского уровня как по количественным, так и по качественным показателям.
Основными задачами Стратегии являются:
1. Увеличение обеспеченности населения, учреждений системы здравоохранения и Вооруженных Сил Российской Федерации, федеральных органов исполнительной власти, в которых законом предусмотрена военная и приравненная к ней служба, жизненно необходимыми и важнейшими лекарственным средствами отечественного производства, а также лекарственными средствами для лечения редких заболеваний;
2. повышение конкурентоспособности отечественной фармацевтической промышленности путем гармонизации российских стандартов по разработке и производству лекарственных средств с международными требованиями;
3. стимулирование разработки и производства инновационных лекарственных средств;
4. защита внутреннего рынка от недобросовестной конкуренции и выравнивание условий доступа на рынок для отечественных и зарубежных производителей;
5. осуществление технологического перевооружения российской фармацевтической отрасли;
6. совершенствование системы подтверждения соответствия качества лекарственных средств, включая меры по устранению избыточных административных барьеров по регистрации отечественных лекарств;
7. подготовка специалистов для разработки и производства фармацевтической продукции в соответствии с международными стандартами.
Стратегия развития национальной фармацевтической промышленности основывается на следующих приоритетах:
− приоритет инновационной модели развития отрасли;
− приоритет качества, эффективности и безопасности лекарственных средств;
− приоритет национальной фармацевтической отрасли в реализации государственных программ в области обеспечения лекарственными средствами;
− приоритет производства высокотехнологичных фармацевтических субстанций на территории РФ;
− приоритет развития экспортоспособных производств и новых разработок; − приоритет замещения импортных лекарственных средств отечественными, полный цикл производства которых находится на территории РФ;
− приоритет фармацевтической продукции, произведенной на территории РФ, в закупках по перечню жизненно необходимых и важнейших лекарственных средств, а также при осуществлении поставок лекарств для Вооруженных Сил Российской Федерации, федеральных органов исполнительной власти, в которых законом предусмотрена военная и приравненная к ней служба.
6.2. Ожидаемые результаты реализации Стратегии
Ожидаемым результатом реализации Стратегии развития фармацевтической промышленности Российской Федерации на период до 2020 года должно стать:
− увеличение доли продукции отечественного производства в общем объеме потребления на внутреннем рынке до 50% в стоимостном выражении к 2020 году;
− изменение номенклатуры производства лекарственных препаратов, произведенных на территории Российской Федерации, в том числе увеличение доли инновационных препаратов1 в портфелях локальных производителей до 60% в стоимостном выражении;
− увеличение экспорта фармацевтической продукции в 8 раз по сравнению с 2008 годом;
− обеспечение лекарственной безопасности Российской Федерации согласно номенклатуре стратегически важных лекарственных средств и вакцин.
− стимулирование организации производства фармацевтических субстанций на территории Российской Федерации в размере, необходимом для обеспечения выпуска 50% готовых лекарственных форм в денежном выражении, включая не менее 85% по номенклатуре из списка стратегических ЛС.
Рисунок 1. Доля рынка продукции отечественного и зарубежного производства в 2011 году и планируемая Стратегией к 2020 году.

6.3. Проблемы фармацевтической отрасли
Следует отметить три системные проблемы российской фармацевтической промышленности.
1. Неспособность обеспечивать население Российской Федерации основной номенклатурой современных лекарственных препаратов, весь цикл производства которых находился бы на территории РФ (неспособность обеспечить в текущий момент времени).
2. Низкий уровень инноваций и технологий, используемых при разработке и производстве ЛС (неспособность обеспечить в «перспективе»). Эта общая проблема российской экономики особенно актуальна для фармацевтического сектора.
3. Низкий уровень обеспечения лекарственной безопасности Российской Федерации, в том числе Вооруженных Сил Российской Федерации, по номенклатуре лекарственных средств, используемых в военное время для оказания медицинской помощи и лечения пораженных ионизирующим излучением и боевыми отравляющими веществами.
Составляющие факторы системных проблем
1. Дисбаланс регуляторных требований к отечественным и зарубежным предприятиям-производителям. Регуляторные процедуры, предусмотренные законодательством для отечественных и иностранных производителей, в целом различны. Например, процедуры регистрации фармацевтических субстанций в настоящее время являются существенно более сложными и длительными для отечественных производителей по сравнению с зарубежными. Так, порядок выдачи лицензий на производство субстанций отечественным производителям предполагает физический контроль всего производственного цикла (а также проведение 20 последующих проверок на соответствие лицензионных требований и условий каждые 2 года), в то время как зарубежным надлежит только ознакомиться с документацией. Следует отметить, что отдельной процедуры регистрации субстанций в странах ЕС и США вообще не существует.
2. Экономическая демотивация отечественных производителей. Высокий уровень инфляции, укрепление рубля, высокие ставки процентов по кредитам, рост тарифов на энергоносители, высокая стоимость капитального строительства в силу географического расположения, демпинговая политика азиатских государств — все эти факторы изначально ставили локального производителя в неравные конкурентные условия с зарубежным и, по экспертным оценкам, снижали конкурентную способность отечественных фармпроизводителей до 50%. Однако вызванное общемировым кризисом ослабление рубля в настоящий момент отчасти снимает данные проблемы и создает предпосылки для более ускоренного импортозамещения. Без учета НДС и подоходного налога на заработную плату российские компании выплачивают по налогам 12—14% от объема продаж; зарубежные компании, не имеющие представительств в России, оплачивают только незначительный таможенный сбор (от 0 до 10%). Система преференций для малого бизнеса в государственных закупках лекарственных средств в отличие от других развитых стран реализуется неэффективно.
3. Дефицит высококвалифицированных кадров для фармацевтической отрасли. Из-за отсутствия масштабного спроса со стороны индустрии, в России практически отсутствует подготовка высококвалифицированных кадров для современного фармпроизводства и индустриальной науки. Налаживание опережающей подготовки и переподготовки таких специалистов является ключевым условием успешной реализации настоящей Стратегии. Эта задача приобретает особую актуальность, учитывая ситуативное, связанное с 21 кризисным периодом, высвобождение трудовых ресурсов, которые могут быть задействованы в фармацевтическом производстве. Одной из важнейших проблем подготовки кадров для отечественной фарминдустрии является чрезвычайно низкий уровень оплаты труда профессорско-преподавательского состава ВУЗов. Все это приводит к оттоку квалифицированных кадров в другие сферы деятельности и за границу, к утрате традиций и уровня преподавания из-за ухода пожилых преподавателей, не имеющих возможности передавать свою квалификацию молодой смене. Серьезной проблемой является недостаточная практическая подготовка выпускников — следствие, как неразвитой материально-технической базы ВУЗов, так и недоработок в содержательном аспекте базовой программы. Эти же проблемы характерны для послевузовского и дополнительного этапов образования. Следствием оттока молодых преподавателей в другие области хозяйства является низкая ориентированность ВУЗов на восприятие к обучению новым направлениям науки и технологии, постоянно появляющимся в мире.
4. Отсутствие механизмов финансирования разработок лекарственных средств. Без наличия достаточного объема высокорентабельных инновационных препаратов в своих продуктовых портфелях российские фармацевтические производители вынуждены конкурировать за счет цены и рекламы, что, соответственно, снижает объемы средств на разработку новой продукции. В сложившейся ситуации российская фарминдустрия не сможет выжить, будучи зажата между западными транснациональными корпорациями, диктующими правила игры в сфере технологий и интеллектуальной собственности, и производителями из Индии и Китая с их беспрецедентным ценовым давлением. Ни зарождающиеся отечественные компании- разработчики лекарств, ни создаваемые венчурные фонды пока не готовы финансировать долгосрочные и рискованные по своей природе разработки 22 инновационных фармацевтических препаратов. Существующий частный бизнес и венчурные инвесторы участвуют в развитии лишь тех подсекторов, которые дают прибыль в короткие сроки и не требуют крупных рискованных инвестиций (аптечные сети, фабрики по производству готовых лекарственных форм, биологически активных добавок и др.). Фактически речь идет о разрывах в критических цепочках взаимодействий, обеспечивающих непрерывное функционирование инновационной системы по созданию новых отечественных препаратов. Более того, недофинансирование ранних стадий разработки новых лекарств в виде грантов и посевного финансирования, приводит к фактическому отсутствию отраслевой науки. Однако, необходимо отметить что идущий мировой кризис создает дополнительные возможности для российской фарминдустрии по трансферу в Россию западных разработок, остановленных из-за недофинансирования, на очень выгодных условиях. Эти возможности могут быть реализованы только при комплексной (финансовой и организационной) поддержке со стороны государства и привлечению ключевых институтов развития, таких как ГК «Роснанотех» и ОАО «РВК» (создание корпоративных венчурных фондов), ГК «Банк развития и внешнеэкономической деятельности (Внешэкономбанк)» (инвестиционное финансирование) и ГК «Ростехнологии» (содействие разработке и производству).
5. Недостаточный уровень российского патентного законодательства и законоприменительной практики относительно международных стандартов. Несмотря на отмечаемые многими экспертами улучшения в области защиты интеллектуальной собственности, российские организации и институты, работающие в этой сфере, пока не готовы адекватно отвечать на требования времени.
6. Отсутствие обязательных для исполнения правил производства и контроля качества лекарственных средств, идентичных международным правилам GMP Несмотря на то, что Госстандартом России утвержден национальный стандарт ГОСТ Р 52249-2004 «Правила производства и контроля качества лекарственных средств» (стандарт является идентичным переводом Правил GMP Европейского союза), до сих пор вопрос об обязательности правил GMP, идентичных международным, остается открытым, что тормозит не только выпуск более качественной продукции для отечественного потребителя, но и ее выход на международные рынки.
Указанные проблемы ведут к тому, что при нынешнем инерционном сценарии развития, несмотря на имеющиеся позитивные предпосылки, включая повышение конкурентоспособности отечественных производителей за счет ослабления рубля, в России в обозримом будущем могут перестать существовать как базовый производственный сектор, производящий лекарственные субстанции, так и сопряженный с ним научно-технологический сектор
6.4. Основные мероприятия и ожидаемые результаты реализации Стратегии
Настоящая Стратегия предусматривает реализацию трех основных этапов:
I этап — «Локализация производства и разработки лекарственных средств на территории РФ».
II этап — «Развитие фармацевтической отрасли на рынке РФ»
III этап — «Развитие фармацевтической отрасли на внешних рынках».
Для большинства мероприятий необходимо проведение предварительных действий, а также последующих мониторинговых шагов после их реализации. Отнесение мероприятий к тому или иному этапу означает проявление максимального внимания в этом интервале и получение соответствующего эффекта.
Рисунок 2. Основные этапы реализации Стратегии.
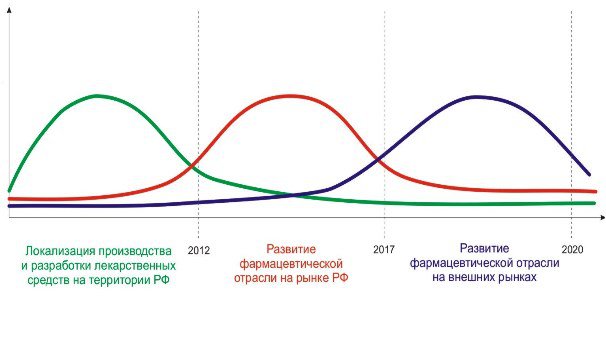
ПЕРВЫЙ ЭТАП: ЛОКАЛИЗАЦИЯ ПРОИЗВОДСТВА И РАЗРАБОТКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ НА ТЕРРИТОРИИ РФ
Основная цель первого этапа заключается в создании системы современного фармацевтического производства и разработки ЛС на территории РФ. Основной задачей является развитие современной производственной базы (в том числе с помощью локализации высокотехнологичных производств и исследовательских центров на территории РФ), соответствующей стандартам GMP, позволяющей с высокой эффективностью производить лекарственные субстанции и готовые лекарственные формы на их основе.
Для реализации первого этапа будут приняты меры, направленные на преодоление основных негативных тенденций в национальной фармацевтической промышленности:
• устранение существующего конкурентного неравенства между локальными и зарубежными фармацевтическими производителями в Российской Федерации, в том числе в нормативно-правовом поле;
• внедрение обязательных требований к правилам производства лекарственных средств (GMP), идентичных международным. Создание инспекции по GMP;
• разработка механизма прямых компенсаций затрат и/или соответствующих налоговых выплат фармацевтическим предприятиям, перешедшим на правила GMP, из средств федерального бюджета в размере до 50% рас ходов (после предъявления документов о соответствии производства требованиям GMP);
• модернизация и утверждение Фармакопеи РФ, гармонизированной с Европейской Фармакопеей;
• исключение требования обязательного утверждения и регистрации Фармакопейных статей предприятия;
• замена требования проведения предрегистрационной экспертизы качества лекарственных препаратов экспертизой в рамках процедуры предварительного государственного контроля качества;
• введение требования предоставления регистрационного досье на лекарственный препарат в формате CTD (Common Technical Document);
• ограничение срока действия регистрационного удостоверения на ЛС пятью годами при первичной регистрации; при последующей перерегистрации — получение бессрочного удостоверения;
• принятие документов, регламентирующих разработку лекарственных средств в соответствии с международными стандартами надлежащей лабораторной и клинической практики (GLP и GCP).
• модернизация системы подготовки высококвалифицированных специалистов в области разработки и производства лекарственных средств;
• разработка и принятие необходимых изменений в законодательстве Российской Федерации и соответствующих нормативно-правовых актов, развитие государственных программ Российской Федерации в области лекарственного обеспечения;
• организация функционирования федеральных лабораторий для осуществления государственного контроля качества и безопасности ЛС;
• введение для производителей ЛС института уполномоченных лиц, отвечающих за качество и безопасность производимой продукции, несущих наравне с руководителем административную и уголовную ответственность за качество и безопасность продукции, выпускаемой предприятием;
• проведение комплекса мероприятий, направленных на борьбу с коррупцией в вопросах лекарственного обеспечения;
• проведение комплекса мероприятий, направленных на обеспечение лекарственной безопасности по лекарственным средствам военного назначения.
Второй этап:развитие фармацевтической отрасли за счет рынка РФ
Основной задачей является формирование эффективного рыночного механизма по высокотехнологичному импорто- замещению лекарственных средств.
Основная цель второго этапа заключается в создании отлаженной системы по производству и выводу на рынок дженериковых ЛС.
Необходимо реализовать две группы специфических мероприятий:
-мероприятия по поддержке отечественных предприятий-производителей
фармацевтических субстанций;
-мероприятия по поддержке отечественных предприятий-производителей готовых лекарственных форм (ГЛФ).
Необходимость разделения этих групп мероприятий связана с наличием в отрасли ярко выраженной системы разделения труда, при которой одна часть предприятий производит лекарственные субстанции, а другая часть — ГЛФ. При этом как практические задачи, так и экономические интересы этих групп зачастую существенно отличаются.
Мероприятия по поддержке отечественных предприятий-производителей фармацевтических субстанций:
− внесение в законодательные акты дополнений, гарантирующих при организации закупок для государственных нужд приоритетность отечественных производителей с целью локализации производств.
− внесение в нормативно-правовое регулирование в сфере обращения лекарственных средств дополнений, выравнивающих требования для отечественных и зарубежных фармпроизводителей в части порядка контроля качества субстанций; для этого предусмотреть аудит роизводителей импортных субстанций государственными экспертными организациями.
Мероприятия по поддержке отечественных предприятий-производителей готовых лекарственных средств:
− продолжение создания и внедрения в практику преференций при участии в конкурсе государственных закупок ЛС для локальных производителей;
− формирование Минздравсоцразвития и Минпромторгом РФ перечня лекарственных средств российского производства, рекомендуемой для государственных и региональных закупок (в т. ч. по программе обеспечения необходимыми лекарственными средствами);
− отмена регистрации субстанций при ужесточении контроля качества ГЛФ, в том числе путем обеспечения выездных инспекций и аккредитации в
регуляторных органах РФ всех зарубежных производителей фармацевтических субстанций и ГЛФ, поступающих на рынок РФ;
− поддержка деятельности отечественных фармацевтический компаний
на внешних фармацевтических рынках, в рамках мер предусмотренных для экспортеров промышленной продукции (стран СНГ, ближнего и дальнего зарубежья);
− отмена требования регистрации ЛС,произведенного исключительно
для экспорта;
− внесение изменений в Постановление Правительства РФ от 16 июля 2005 года
№438, разрешающих ввоз зарегистрированных активных фармацевтических субстанций предприятием-производителем лекарственных средств для собственного производства на основании регистрационных удостоверений на лекарственный препарат и активную фармацевтическую субстанцию.
Третий этап: развитие фармацевтической отрасли за счет внешних рынков
Третья группа мероприятий предусматривает реализацию мер, направленных на развитие конкурентных преимуществ национальной фармацевтической отрасли и осуществление ее перехода на инновационную модель развития.
Основная задачасостоит в создании инфраструктуры для разработки инновационных препаратов с применением последних достижений науки и техники и использовании современных технологических платформ.
Основными целями данных мероприятий являются:
· разработка и производство отечественных инновационных препаратов для импортозамещения лекарственных средств, находящихся под патентом на локальном рынке.
· Разработка и производство по актуальным фармацевтическим направлениям отечественных патентоспособных препаратов, которые имеют известные зарубежные прототипы и показали клиническую эффективность. Фактически речь идет об оригинальных лекарственных препаратах, имеющих тот же механизм действия и не меньшую эффективность, чем зарубежные прототипы, но обладающих дополнительными полезными свойствами, такими как пониженная токсичность, повышенная биодоступность и т.п.
Специфические мероприятия:
− поддержка НИР и НИОКР, направленных на создание импортозамещающих ЛС и ГЛФ, в том числе путем использования потенциала государственных научных учреждений и регулярного проведения конкурсов среди малых научных фирм на разработку новых ЛС с последующей приоритетной закупкой государством созданных препаратов;
− выделение в рамках государственных институтов развития средств на создание специализированных финансовых инструментов, в том числе корпоративных и посевных фондов, для финансирования разработки инновационных лекарственных препаратов и фармацевтических субстанций, а также приоритетное финансирование проектов по разработке импортозамещающих лекарственных препаратов по направлению «Живые системы» Федеральным агентством по науке и инновациям (Роснауки) в рамках действующих и будущих ФЦП;
− стимулирование внедрения современных исследовательских технологий, в том числе на основе высокопроизводительных лабораторных испытаний;
− стимулирование эффективных патентных исследований и мониторинга международных рынков;
− совершенствование процедур, регламентирующих проведение доклинических и клинических испытаний;
− стимулирование нанобиотехнологий для решения вопросов эффективной доставки в организм человека известных ЛС и создания инновационных ГЛФ;
− создание системы выделения грантов для малых научных предприятий,
связанных с разработкой лекарств;
− создание масштабной программы поддержки кооперационных проектов, объединяющих организации разных форм собственности и отраслевой принадлежности, работающие в сфере разработки и производства ЛС и ГЛФ;
− формирование спроса и стимулирование подготовки исследовательских кадров нового поколения, в том числе с участием государственных академий наук.
Разработка и производство отечественных инновационных препаратов, экспортоспособных на мировых рынках.
Результатом реализации этого комплекса мероприятий в рамках настоящей Стратегии станет появление к 2020 году значительного числа отечественных разработок, позволяющих наладить реализацию готовой продукции или получение лицензий на ее производство за рубежом. Существенным отличием инновационных препаратов от патентоспособных структурно-модифицированных аналогов является реализация полного цикла разработки лекарств с нуля с использованием передовых научно-исследовательских подходов. Только такой подход позволяет в полной мере реализовать достижения «геномной эры» и создавать препараты нового поколения, отличающиеся высокой эффективностью, низким уровнем побочных эффектов и высокой рентабельностью производств.[13]
6. 5. Объем и источники финансирования мероприятий Стратегии
Источниками финансирования расходов на реализацию мероприятий Стратегии развития фармацевтической промышленности Российской Федерации на период до 2020 года являются федеральный бюджет, средства коммерческих и общественных организаций и иные внебюджетные средства. Важными механизмами финансирования разработки новых лекарственных средств на этапе доклинических испытаний и при прохождении первой и второй фаз клинических испытаний должны стать существующие академические учреждения, венчурные фонды и фонды посевного финансирования. Следует также осуществлять финансирование таких проектов в рамках федеральных целевых программ (ФЦП) и необходимо использовать имеющиеся возможности банков по выделению кредитов, в том числе для перевода отраслевых производств на стандарты GMP, а также стимулировать фармпроизводителей к инвестиционной активности в области разработки новых лекарств, в том числе за счет созданий корпоративных венчурных фондов. Общая схема источников финансирования до 2017–2018 годов представлена на рис.3.
Рисунок 3. Источники инвестирования в рискованные проекты разработки новых лекарств.
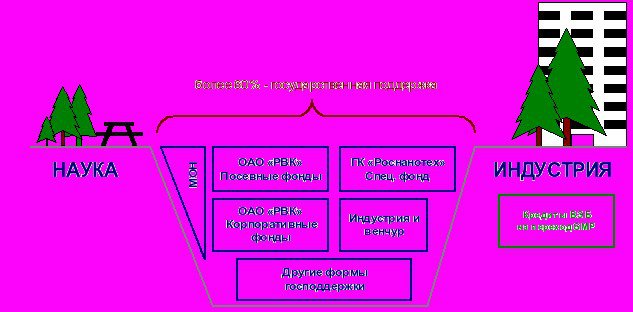
На первом этапе запуска «отраслевого инновационного цикла» фарминдустрии потребуется до 75% от всех средств со стороны государственных источников. Государственное финансирование впоследствии будет полностью замещено внебюджетными источниками со стороны фарминдустрии и венчурных фондов на этапе коммерциализации ранее разработанной интеллектуальной собственности. При этом,дальнейшее финансирование новых разработок в отрасли будет осуществляться уже за счет внебюджетных источников, так как значительный объем инновационных препаратов в портфелях отечественных производителей позволит им выделять до 15% своего годового оборота новые разработки, как это принято в развитых странах.
Объем финансирования был рассчитан исходя из основной задачи Стратегии по достижению отечественной промышленностью к 2020 году не менее 50% доли рынка в денежном выражении. Решением этой задачи является ком- плекс мер, в том числе финансовое стимулирование дженерикового импортозамещения и разработки отечественных инновационных лекарственных средств. В рамках решения этой задачи необходимо также финансовое стимулирование перехода отрасли на стандарты GMP. Фармацевтическая индустрия, являющаяся одним из важнейших элементов системы здравоохранения, стоит на пороге коренных изменений. В наибольшей степени эти изменения должны быть связаны с формированием инновационной составляющей, развитием импортозамещения и ростом производительности труда. Инновационный сценарий развития событий предполагает разработку и принятие Стратегии развития фармацевтической промышленности России, призванной решить проблему лекарственного обеспечения населения России в существующих условиях и на долгосрочную перспективу. Конечной целью всех этих инициатив является создание устойчивой национальной индустрии, способной обеспечить население Российской Федерации доступными, эффективными и безопасными лекарствами в необходимых количествах. Важнейшим элементом Стратегии должна быть направленность на создание нового поколения инновационных лекарств. Настоящая Стратегия отражает стратегические цели, принципы и задачи развития фармацевтической промышленности, ситуацию в отрасли, проблемы отрасли, способы и пути решения указанных проблем.[12]
7.Исследование доли выпускаемой продукции отечественных производителей в ассортименте товаров аптек базы практики.
7.1.Анализ доли препаратов ЗАО «Эвалар»
В Аптеке ООО «Сатурн», расположенной по адресу: Республика Марий Эл, пгт. Медведево, ул. Юбилейная 3а, имеются лекарственные препараты и БАДы отечественных производителей, имеющих сертификат соответствия правилам GMP:
| Товары | ЗАО «Эвалар» | ОАО «Нижфарм» | «Биокад ООО» | ЗАО «Фарм Фирма «Сотекс» | ОАО ХФК «Акрихин» |
| Лекарст венные препараты | АкваМастер Гинкоум Ци-клим | Анестезол Левоме коль Гексикон Омарон Тагиста Витапрост Дикловит | Генферон лайт | Амелотекс Нейрокс Хондро гард | Унидерм Акридерм ГК Микозорал Ацикловир Капотен Азелик |
| БАДы | Атероклефит Гинго билоба Гепатрин Глицин Форте Кардиоактив МКЦ Лора Пустырник Форте Овесол Олиджим Стиммунал Фитолакс Формула сна Эндокринол | Нормобакт |
Рассчитаем какую долю препараты одного конкретного производителя занимают в общем объеме лекарственных препаратов в целом по аптеке.
В Аптеке ООО «Сатурн» 3 лекарственных препарата ЗАО «Эвалар», общее количество лекарственных препаратов, реализуемых данной аптечной организацией 3598 торговых наименований.
Доля препаратов ЗАО «Эвалар» занимает:
3*100%/3598=0,08%
Проведем анализ спроса на препараты этого производителя и других зарубежных производителей в рамках фармакологичесских групп:
| Фирмы -производители | Ангиопротекторное средство растительного происхождения, корректор мозгового кровообращения | Средство для лечения заболеваний носа, антиконгестанты | Противо климактерическое средство растительного происхождения |
| ЗАО «Эвалар» | Гинкоум | АкваМастер | Ци-клим |
| Другие зарубежные производители | Билобил форте Танакан Мемоплант | Аква Марис Аква Марис Плюс | Климактоплан Климонорм |
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ООО «Сатурн» было реализовано:
Гинкоум капс, 40 мг №30-2 уп.
Танакан таб, 40 мг №30-1 уп.
Билобил форте капс, 80 мг №60-2 уп.
________________________________
Итого: 5 упаковок.
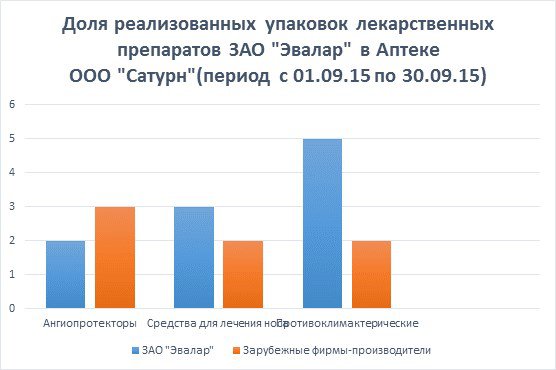
Доля реализованных упаковок препарата «Гинкоум» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Гинкоум - 2*100%/5=40,0%
Танакан - 1*100%/5=20,0%
Билобил форте - 2*100%/5=40,0%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, улучшающих мозговое кровообращение, препарат отечественного производства Гинкоум пользуется спросом наравне с зарубежными препаратами аналогичной группы, так как по эффективности нисколько не уступает им ни по качеству, ни по цене.
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ООО «Сатурн» было реализовано:
Аква Мастер спрей наз, 0,65% 50 мл - 3 уп.
Аква Марис спрей наз, 30 мл -2 уп.
Аква Марис плюс, спрей наз, 30 мл -0 уп.
______________________________
Итого: 5 упаковок.
Доля реализованных упаковок препарата «Аква Мастер» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Аква Мастер – 3*100%/5=60%
Аква Марис – 2*100%/5=40%
Аква Марис Плюс - 0%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, используемых для лечения носа, препарат отечественного производства Аква Мастер пользуется значительным спросом, чем аналогичные зарубежные препараты, так как за эту же цену клиент получает в два раза больше препарата.
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ООО «Сатурн» было реализовано:
Ци-клим таб, №60 -5 уп.
Климактоплан таб, №100 - 1 уп.
Климонорм драже, №21 -1 уп.
__________________________________
Итого: 7 упаковок.
Доля реализованных упаковок препарата «Ци-клим» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Ци-клим - 5*100%/7=71,0%
Климактоплан - 1*100%/7=14,3%
Климонорм -1*100%/7=14,3%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов противоклимактерического действия, препарат отечественного производства Ци-клим пользуется значительным спросом в сравнении с такими же аналогичными препаратами, так как имеет более натуральный состав и более низкую цену.
7.2. Анализ доли препаратов ОАО «Нижфарм»
В Аптеке ИП Козлова, г.Уфа, 13 лекарственных препаратов ОАО «Нижфарм», общее количество лекарственных препаратов, реализуемых данной аптечной организацией 3016 торговых наименований.
Доля препаратов ОАО «Нижфарм» занимает:
13*100%/3016=0,43%
Проведем анализ спроса на препараты этого производителя и других зарубежных производителей в рамках фармакологичесских групп:
| Фирмы- производители | Обезбаливающие и противо воспалительные препараты | Противогем мороидальные средства | Средства, улучшающие мозговое кровооб ращение |
| ОАО «Нижфарм» | Матарен плюс | Натальсид | Омарон |
| Другие зарубежные производители | Найз Нимулид Вольтарен | Прокто-гливенол Релиф Ультрапрокт | Фезам Пирацезин |
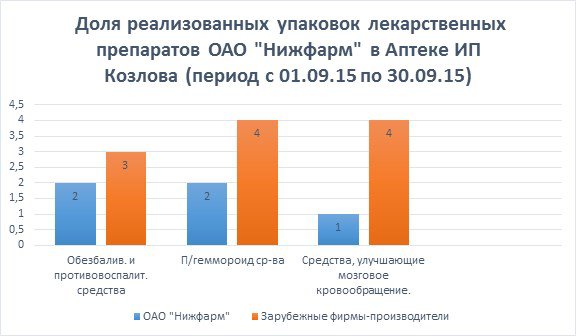
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ИП Козлова было реализовано:
Матарен плюс, крем, 30г -2 уп.
Найз гель, 1% 20г -1 уп.
Нимулид гель, 1% 30г -2 уп.
________________________________
Итого: 5 упаковок.
Доля реализованных упаковок препарата «Матарен плюс» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Матарен плюс - 2*100%/5=40,0%
Найз - 1*100%/5=20,0%
Нимулид - 2*100%/5=40,0%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, обладающих противовоспалительным и обезбаливающим действием, препарат отечественного производства Матарен плюс пользуется таким же спросом, что и аналогичные зарубежные препараты, так как по эффективности нисколько им не уступает и более экономичный по цене.
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ИП Козлова было реализовано:
Натальсид ,супп. 0,25 г №10 - 2 уп.
Релиф, супп.рект. №12 -3 уп.
Прокто-гливенол, супп.рект. №10 – 1 уп.
______________________________
Итого: 6 упаковок.
Доля реализованных упаковок препарата «Натальсид» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Натальсид -2*100%/6=33,3%
Релиф – 3*100%/6=50%
Прокто-гливенол – 1*100%/6=16,6%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, используемых для лечения геммороя, препарат отечественного производства Натальсид пользуется таким же спросом, как и препараты зарубежных фирм-производителей, так как имеет минимальное количество побочных эффектов.
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ИП Козлова было реализовано:
Омарон таб, №30 -1 уп.
Фезам капс, №60 - 3 уп.
Пирацезин капс, №60 -1 уп.
__________________________________
Итого: 5 упаковок.
Доля реализованных упаковок препарата «Омарон» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Омарон - 1*100%/5=20,0%
Фезам - 3*100%/5=60,0%
Пирацезин -1*100%/5=20,0%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, улучшающих мозговое кровообращение, препарат отечественного производства Омарон пользуется незначительным спросом, так как люди мало осведомлены по поводу применения данного препарата.
7.3. Анализ доли препаратов фирмы ОАО ХФК «Акрихин»
В Аптеке ООО «Сазонов И.А.» 19 лекарственных препаратов ЗАО ХФК «Акрихин», общее количество лекарственных препаратов, реализуемых данной аптечной организацией 4230 торговых наименований.
Доля препаратов ОАО ХФК «Акрихин» занимает:
19*100%/4230=0,45%
Проведем анализ спроса на препараты этого производителя и других зарубежных производителей в рамках фармакологических групп:
| Фирмы-производители | Дерматологи ческие средства | Противогриб ковые средства | Средства, применяемые при хрон.венозной недостаточности |
| ОАО ХФК «Акрихин» | Унидерм | Микозорал | Венолайф |
| Другие зарубежные производители | Момат Элоком Адвантан | Низорал Себозол Перхотал | Лиотон Троксерутин Троксевазин |
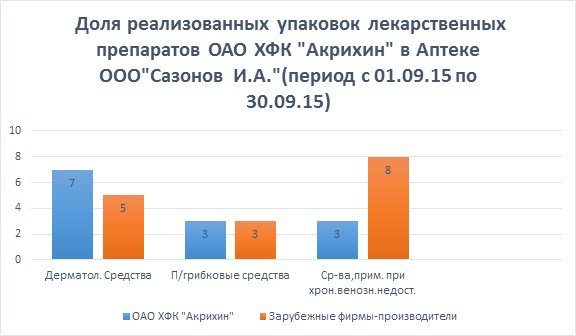
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ООО «Сазонов И.А.» было реализовано:
Унидерм, крем,0,1% 15г -7 уп.
Момат, крем, 0,1% 15г -2 уп.
Элоком, мазь,0,1% 15г -3 уп.
________________________________
Итого: 12 упаковок.
Доля реализованных упаковок препарата «Унидерм» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Унидерм - 7*100%/12=46,6%
Момат - 2*100%/12=16,7%
Элоком - 3*100%/12=25%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, препарат отечественного производства Унидерм пользуется значительным спросом по сравнению с аналогичными зарубежными препаратами, так как более доступен по цене и эффективен.
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ООО «Сазонов И.А.» было реализовано:
Микозорал, шампунь, 2% 60 мл - 3 уп.
Низорал, шампунь,20 мг/г 60 мл -3 уп.
Перхотал, шампунь,1% 60 мл – 0 уп.
______________________________
Итого: 6 упаковок.
Доля реализованных упаковок препарата «Микозорал» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Микозорал - 3*100%/6=50%
Низорал - 3*100%/6=50%
Перхотал - 0%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, препарат отечественного производства Микозорал пользуется спросом наравне с аналогичными зарубежными производителями, так как не уступает по качеству и имеет более низкую цену.
За анализируемый период времени (с 01.09.15г по 30.09.15г) в Аптеке ООО «Сазонов И.А.» было реализовано:
Венолайф, гель, 40г -3 уп.
Лиотон, гель, 30г - 1 уп.
Троксевазин, гель,2% 40г -7 уп.
__________________________________
Итого: 11 упаковок.
Доля реализованных упаковок препарата «Венолайф» по отношению к другим лекарственным препаратам из данной фармакологической группы зарубежных фирм-производителей составляет:
Венолайф - 3*100%/11=27,3%
Лиотон - 1*100%/11=9,09%
Троксевазин-7*100%/11=63,6%
По результатам данного исследования можно сделать вывод, что среди лекарственных препаратов, применяемых при хронической венозной недостаточности, препарат отечественного производства «Венолайф» пользуется спросом, так как имеет более богатый состав по сравнению с зарубежными аналогами.
Заключение
Исходя из вышеизложенного материала можно сделать следующие выводы:
Ø термин надлежащей производственной практики содержит свод необходимых минимальных требований к организации производства фармацевтической продукции и изделий медицинского назначения, направленные на обеспечение их качества. Правила GMP – это комплексная система требований к организации производства и контроля качества фармацевтических средств;
Ø описана история возникновения правил GMP в зарубежных странах и в России. Первоначально правила GMP разрабатывались в каждой стране отдельно, большего успеха в этом достигли США и Великобритании. В России переход к правилам начался с 90-х годов прошлого столетия и только в 2009 г. был принят ГОСТ Р 52249-2009, который является идентичным переводом правил ЕС от 2009 года;
Ø к документам, относящимся к правилам, относятся Стандарт ГОСТ Р 52249-2009, Правила организации производства и контроля качества лекарственных средств были приказом Минпромторга России 14 июня 2013 года №916;
Ø правила GMP основаны на следующих принципах:
• системный подход;
• профилактическая направленность;
• гибкость в способах выполнения требований;
• обязанность производителя представить доказательства адекватности избранного им метода выполнения требований (валидация).
Главной целью внедрения GMP является гарантирование соответствия состава лекарственного средства, отсутствие в нем различных загрязнений, корректность маркировки, неизменность и стабильность свойств в процессе хранения в течении установленного срока годности.
Правила GMP тесно связаны с другими стандартами такими как GLP-Надлежащая лабораторная практика, GSP - Надлежащая практика хранения фармацевтической продукции, GPP - Надлежащая аптечная практика, GDP - Надлежащая дистрибьюторская практика;
Ø с 1 января 2014 года лицензии на производство ЛС выдаются только при условии соответствия правилам GMP, а предприятия, уже получившие бессрочные лицензии, должны их подтвердить, пройдя государственную инспекцию. Этот срок был установлен еще в 2010 году Федеральным законом № 61 «Об обращении ЛС». В России были получены сертификаты соответствия следующими фармацевтическими предприятиями: Компания «Нижфарм» (STADA CIS), ЗАО «Эвалар», ОАО «Фармстандарт-Уфавита», «Озон», ЗАО «ФармФирма Сотекс», Компания «Зио-Здоровье», Компания «Канон-Фарма Продакшн», ОАО ХФК «Акрихин»;
Ø на данный момент гармонизация фармацевтического производства в России с правилами GMP находится в периоде становления и уже получены первые сертификаты соответствия, разработанные и утвержденные Минпромторгом России. Большинство Российских предприятий отстает с внедрением правил GMP на несколько десятилетий от уровня индустриальных стран. В качестве причины запаздывания официально признается только одна – недостаток финансовых средств.
Ø Ассортимент отечественных лекарственных препаратов в Аптеках базы практики увеличивается с каждым годом
Ø Рассчитав долю лекарственных препаратов отечественных производителей и проанализировав спрос на них мы убедились, что наши российские препараты нисколько не уступают зарубежным аналогам, иногда даже превосходя их
Таким образом, исследуя доли выпускаемой продукции отечественных производителей в ассортименте товаров аптек базы практики мы видим тенденцию к увеличению ассортимента лекарственных препаратов на полках аптек и люди проявляют заметный интерес именно к товарам отечественного производства, чувствуется доверие к препаратам, что обусловлено внедрением на фармацевтических предприятиях стандартов GMP- гарантии производства качественной и безопасной продукции, отвечающим международным нормам.
Список использованных источников
1. ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств»
2. К 50-летию GMP: из истории Правил GMP (сообщение первое) /А.П. Мешковский [и др.] // Ремедиум. – 2013. – № 2. – С. 32-39.
3. К 50-летию GMP: изменение подходов к регулированию лекарственного рынка (продолжение) /А.П. Мешковский [и др.] // Ремедиум. – 2013. – № 3. – С.24-26.
4. Мешковский А.П. Что такое GMP. /А.П. Мешковский, Ю.Ф. Крылов //Мнения // Фарматека. - № 2. - С.7-10.
5. Мешковский А.П. О внедрении правил GMP в России /А.П. Мешковский, В.П. Грачев // Фарматека. - № 1. - С.46-50.
6. Организация и контроль производства лекарственных средств. Стерильные лекарственные средства // Методические указания МУ 42-51-1-93 + МУ 42-51-26-93. - Москва, 1993. - 75 с.
7. ОСТ 42-510-98 «Правила организации производства и контроля качества лекарственных средств (GMP)».
8. Правила организации производства и контроля качества лекарственных средств // Руководящий нормативный документ РД 64-125-91. - Москва, 1991. - 50 с.
9. Санитарные правила СП 3.3.2015-94 «Производство и контроль медицинских иммунобиологических препаратов для обеспечения их качества».
10. Федотов А.Е. Основы GMP: производство лекарственных средств // Издательство: M.: АСИНКОМ.-2012.-583 c.
11. Федеральный закон Российской Федерации «О лекарственных средствах» // Фармация. - 1998 - № 4. - С.3-17.
12.Стратегия развития фармацевтической промышленности Российской Федерации на период до 2020 года [Электронный ресурс]. —
режим доступа: http://pharma2020.ru/
13. Соколов И.Б., Лин А.А, Орлов А.С. Фармацевтический рынок: структурные особенности в России. // Проблемы современной экономики. — 2012. — №4 (44).- С. 336-341.
0 комментариев