Кратко рассмотрены сведения о программируемой клеточной смерти (апоптозе) у животных и растений. Патологический вариант клеточной гибели – некроз, сопровождаемый у животных воспалительным процессом. В результате апоптоза клетки животных и растений дробятся на мембранные везикулы с внутриклеточным содержимым. Эти везикулы у животных поглощаются соседними или специализированными клетками (фагоцитами). У растений нет таких специализированных клеток, фагоцитозу препятствуетналичие клеточной стенки. Рассмотрены основные молекулярные механизмы апоптоза у животных, пути активации каспаз – эволюционно-консервативных цистеиновых протеаз. В особый раздел выделены процессы программируемой клеточной смерти у микроорганизмов: 1)гибель клеток слизистого гриба Dictyostelium discoideum и паразитического жгутиконосца Trypanosoma cruzi; 2)программируемая гибель генноинженерных штаммов дрожжей, экспрессирущих проапоптозные белки Bax и Bak; 3)гибель части клеточной популяции прокариот при исчерпании питательного субстрата или под влиянием стресс-факторов; 4)элиминация клеток Escherichia coli, лишившихся плазмидных систем, кодирующих стабильный цитотоксический агент в сочетании с лабильным противоядием к нему; 5) программируемая гибель бактериальных клеток, зараженных фагом. Обсуждается взаимосвязь программируемой смерти и некультивируемого состояния у микроорганизмов.
This paper briefly overviews the data on programmed cell death (apoptosis) in animals and plants. Necrosis represents a pathological scenario of cell death, which is accompanied by inflammation in animals. Apoptosis results in disintegration of animal and plant cells into membrane vesicles enclosing the intracellular content, which are thereupon absorbed by adjacent cells or specialized cells (phagocytes). Plants lack such specialized cells, and plant cell walls prevent phagocytosis. The paper considers the main molecular mechanisms of apoptosis in animals and the pathways of activation of caspases, highly conserved cysteine proteases. A special section concerns itself with the process of programmed cell death (PCD) in microorganisms including (i) cell death in the myxomycete Dictyostelium discoideum and the parasirtic flagellate Trypanosoma cruzi; (ii) PCD in genetically manipulated yeasts expressing the proapoptotic proteins Bax and Bak; (iii) death of part of a prokaryotic cell population upon depletion of nutrient resources or under stress; (iv) elimination of cells after a loss of plasmids encoding a stable cytotoxic agent in combination with a labile antidote; (v) PCD in phage-infected bacterial cells.
У многоклеточных организмов – животных, растений и грибов – генетически заложена программа гибели клеток. Формообразовательные процессы в онтогенезе, позитивная и негативная селекция Т- и В-лимфоцитов у животных, гиперчувствительный ответ растений на вторжение патогена, осенний листопад – лишь несколько примеров программируемой клеточной смерти (ПКС). ПКС способствует сохранению порядка и нормального функционирования биологической системы, очищая от невостребованных, больных, закончивших свой жизненный цикл или появившихся врезультате мутаций потенциально опасных клеток. Количество научных работ в этой области нарастает по экспоненте. Цель настоящей работы – кратко рассмотреть сведения об апоптозе у животных и растений и затем, базируясь на этих данных, ответить на вопрос, существует ли программируемая гибель клеток у микроорганизмов.
Апоптоз и некроз – два варианта клеточной смерти
Клеточная смерть известна с момента открытия самой клетки, еще с 1665 г.: Р. Гук (R. Hooke) (см. [1]) описал формации коры дуба из погибших клеток. Однако долгое время это наблюдение оставалось без внимания. Первые гистологические описания клеточной смерти опубликовали К.Фогт (C.Vogt) в 1842 г. (см. [2]) и Р. Вирхов (R.Virchow) в 1859 г. (см. [3]). В формировании современных представлений о ПКСважное место занимает работа Kerr et al. [4] о существовании двух различных видов клеточной смерти у животных – апоптоза и некроза.
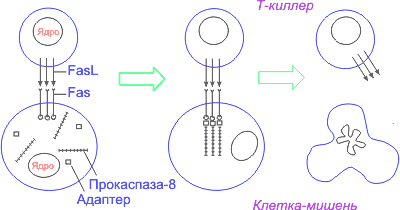
Вот пример апоптоза из области иммунологии. Вирусы, некоторые бактерии, грибы и простейшие являются внутриклеточными паразитами. Хотя специфичные к ним антитела вырабатываются организмом человека или животного, они не могут настигнуть вредителя, затаившегося в цитоплазме, под покровом клеточной стенки жертвы. И тогда в ход вступают цитотоксические Т-лимфоциты (Т-киллеры). Они убивают клетки, ставшие жертвами инфекционного возбудителя, и тем самым прекращают его дальнейшее размножение. Обладая аппаратом распознавания зараженной клетки (клетки-мишени) среди массы здоровых клеток, Т-киллер вызывает ее гибель, включая программу самоубийства клетки-мишени – генетически запрограммированную клеточную смерть, или апоптоз.
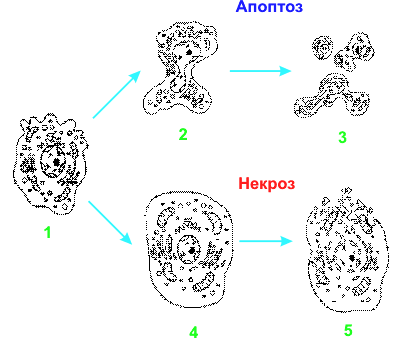
Картина апоптоза у животных – это переход фосфатидилсерина из внутреннего монослоя цитоплазматической мембраны в наружный монослой, уменьшение объема клетки, сморщивание цитоплазматической мембраны, конденсация ядра, разрывы нити ядерной ДНК и последующий распад ядра на части, фрагментация клетки на мембранные везикулы с внутриклеточным содержимым (апоптозные тельца), фагоцитирующиеся макрофагами и клетками-соседями. Такая же участь постигает клетку, когда в ней произошла мутация, которая может привести к опухолевому разрастанию ткани, когда она становится ненужной для организма, например, в процессе онтогенетического развития или, применительно к лимфоцитам, на заключительных этапах инфекционного процесса, когда организм уже не нуждается в дальнейшей выработке антител [5–7].
Есть и другая, патологическая, форма клеточной смерти – некроз. Такая смерть постигает клетку, когда Т-киллер своевременно не распорядился судьбой инфицированной клетки, наставив ее на путь апоптоза. Вирус или иной паразит, размножившись в клетке, разрушает ее: клетка лизируется, ее содержимое изливается наружу, в межклеточное пространство. Некоторые внутриклеточные паразиты, включая простейшее Toxoplasma gondii (возбудитель токсоплазмоза), способны к подавлению апоптоза [8]. Новое поколение паразитов устремляется в соседние клетки, нанося все больший и больший ущерб организму. Начинается воспалительный процесс, исходом которого может быть как выздоровление, так и гибель организма. Некротическую гибель могут вызывать физические или химические повреждения, например, обморожение или ожог, органические растворители, гипоксия, отравление, гипотонический шок и др. [5, 7]. Воспаление – зачастую это катастрофа для окружающих клеток в организме животного.
Симптомы воспаления сформулированы еще А.К.Цельсом (A.C.Celsus) – это "rubor, calor, tumor et dolor" (покраснение, жар, опухание и боль) [9]. Все это в конечном итоге может привести к нарушению функции (functio laesae) и даже к гибели организма. Галанкин и Токмаков [9] сравнивают воспаление с военными действиями на уровне государств: "В мирное время государство… (многоточие в цитате означает пропуск слов оригинального текста) живет в условиях сбалансированной экономики… Его военный потенциал достаточен для выполнения доктрины разумного сдерживания потенциального противника… Но вот… возникает военный конфликт. Цель освобождения территории от неприятеля достигается … не без потерь: экономика перестраивается…, взрываются коммуникации и трубопроводы, разрушаются жилища и мосты, рвутся линии электропередач, при отступлении применяется тактика выжженной земли… Предположим, армия наступает. Далеко не каждая пуля, выпущенная бегущей пехотой, точно попадает в цель, в спешке моторизованные части могут перевернуть понтон, оглушенный телефонист может неправильно понять приказ, авиация – выбросить бомбу на собственную автоколонну и т.д.". Элементы разрушительного характера являются атрибутами воспалительной реакции. Наличиеили отсутствие воспаления у животных используется как признак, позволяющий отличить апоптоз от некроза.
Некроз характеризуется разрывом цитоплазматической и внутриклеточных мембран, что приводит к разрушению органелл, высвобождению лизосомальных ферментов и выходу содержимого цитоплазмы в межклеточное пространство (рис. 1). При апоптозе сохраняется целостность мембран, органеллы выглядят морфологически интактными, а продукты дробления клетки, апоптозные тельца (или везикулы) представляют собой отдельные фрагменты, окруженные мембраной (рис. 1).
Форма клеточной гибели – по пути апоптоза или некроза – во многом определяется внутриклеточной концентрацией NAD+ и АТР. Снижение уровня NAD+ [11] и АТР [12–14] ведет к индукции некроза.
В нормальном организме ПКС – механизм для поддержания гомеостаза. Как гипофункция, так и гиперфункция апоптоза ведут к нарушению гомеостаза.
Итак, ПКС – естественный этап в жизнедеятельности клеток животных. А как дело обстоит у растений? С физиологической клеточной смертью в жизни растений связаны процессы формообразования в онтогенезе, иммунные реакции на внедрение патогена (см. обзоры [1, 15–22]).
Ксилогенез и флоэмогенез. Клетки, которым предстоит стать трахеидами ксилемы, выполняющей водопроводящую и опорную функцию, претерпевают дробление протопласта на везикулы, характерные для апоптоза животных [1, 17, 19, 20]. Развитие ситовидных трубок флоэмы тоже сопровождается дроблением ядра, как при апоптозе. Показано существование генов ted2, ответственных за реализацию процесса сосудообразования [1].
Формообразование листьев. Очертания листьев у растений, по-видимому, формируются через механизм ПКС. У представителей р. Monstera (сем. Аронниковые), места пеpфораций, наличие лопастей определяется зонами гибели клеток на ранних стадиях развития. Этот процесс также находится под генетическим контролем [1, 17].
Аэренхимогенез. Это адаптивная реакция растений на дефицит кислорода, например, при затоплении, заключающаяся в образовании полостей, заполненных воздухом, за счет элиминации некоторых клеток с полным разрушением клеточных стенок с участием активирующихся гидролитических ферментов [15, 17].
Клетки корневого чехлика. Эти клетки защищают апикальную меристему корня при прорастании семян [17]. Программа гибели этих клеток включается даже при выращивании растений гидропонным способом.
Опадание листьев и созревших плодов [1, 17, 22]. Эти процессы сопровождаются избирательной гибелью клеток отделительной зоны, расположенной между основанием черешка листа или плода и стеблем, которая активируется благодаря экспрессии так называемых sag-генов (senescence-associated genes). Клетки в отделительном слое секретируют ферменты, разрушающие клеточные стенки (пектиназы и целлюлазы). Локально действуя на определенный участок, ферменты частично растворяют клеточную стенку в отделительном слое, а сами клетки отделительного слоя подвергаются автолизу, клеточные полимеры распадаются. Одновременно с этим в слое клеток со стороны стебля откладывается водоустойчивый суберин, защищающий оголенный участок ткани, возникающий после отделения листа в результате ферментативного гидролиза полимеров.
Прорастание пыльцевой трубки. Этот процесс осуществляется в результате гибели клеток на пути прорастающей пыльцевой трубки, зависит от видовой принадлежности пыльцы и не включается при действии чужеродной пыльцы [1].
Алейроновые клетки. В семенах однодольных растений имеются алейроновые клетки. При прорастании семян эти клетки секретируют ферменты, катализирующие гидролиз запасных полимеров эндосперма и обеспечивающие тем самым питание проростка. Будучи ненужными для последующего развития, алейроновые клетки погибают по завершении прорастания. По ряду признаков их гибель имеет характер ПКС [17]. Ядерная ДНК расщепляется на олигонуклеосомальные фрагменты, процесс стимулируется гиббереловой и блокируется абсцизовой кислотой (см. [17]). В расщеплении ДНК принимают участие нуклеазы, активность которых регулируется гиббереловой и абсцизовой кислотой [23].
Соматический эмбриогенез [17]. В суспензионных культурах клеток некоторых видов растений индуцируется соматический эмбриогенез: из одиночных клеток можно вырастить взрослые растения – свойство тотипотентности растений. Тотипотентные клетки делятся асимметрично на две клетки, одна из которых останавливает синтез ДНК и погибает спризнаками ПКС, тогда как другая, продолжая развиваться, дает проросток.
Иммунная реакция на патоген [1, 16–18, 21, 24–26]. Устойчивость растения к патогену определяется способностью распознать и своевременно включить механизм защиты. Наряду с индукцией синтеза фитоалексинов (растительных антибиотиков) и гидролитических ферментов, к таким механизмам относится активация в инфицированных клетках и клетках, локализованных вблизи очага инфекции, программы собственной гибели – процесс, называемый гиперчувствительным ответом (ГО). Образуется зона мертвых обезвоженных клеток, которая служит барьером для дальнейшего распространения патогена. ГО – ответ растений на патогенные вирусы, бактерии, грибы и нематоды. При этом гибель клеток растений вызвана не прямым деструктивным действием патогена, а активацией патогеном генетической программы гибели растительной клетки. Процесс сопровождается дыхательным взрывом – генерацией О2Aпри участии NADPH-оксидазы цитоплазматической мембраны, подобной таковой у макрофагов и нейтрофилов млекопитающих. Кроме NADPH-оксидазы, Н2О2 в клетках растений образуется при участии пероксидаз клеточной стенки [27], оксалатоксидазы [28], а также при окислении NAD+-зависимых субстратов или сукцината комплексами I или II дыхательной цепи митохондрий [29]. ГО на одном или нескольких листьях растения ведет к развитию иммунитета в других листьях, которые не имели контакта с патогеном. Такой иммунитет у растений называют системной приобретенной устойчивостью.
Изменения в морфологии клеток растений, претерпевающих ПКС, сходны с таковыми при апоптозе у животных [16, 23, 30]. Также наблюдается конденсация и дробление ядра на фрагменты, съеживание протопласта и складчатость цитоплазматической мембраны. Происходит разрыв плазмодесм – мембранных мостиков, сообщающих содержимое соседних клеток, по которым происходит распространение инфекции из зараженной клетки в соседние [31]. Сжатие протопласта приводит к отделению его от клеточной стенки. В конечном итоге протопласт дробится на отдельные везикулы, аналогичные апоптозным тельцам.
Тогда как у животных апоптозные везикулы in vivo поглощаются соседними или специализированными клетками, у растений нет таких специализированных клеток. Более того, фагоцитозу препятствует наличие клеточной стенки. Апоптозные везикулы у растений в дальнейшем разрушаются. Такая деградация может стимулироваться ферментами, секретируемыми соседними клетками. И в конечном итоге останки разрушенных клеток утилизируются другими клетками, что отмечается при отмирании листьев [22]. При отмирании листа клеточные полимеры распадаются, а мономеры вновь используются растением. Однако такой вариант заключительного этапа клеточной гибели невозможен при ГО, поскольку отмершие клетки несут в себе патоген. В этом случае образуются так называемые демаркационные, отторгающие, ткани вследствие приобретения здоровыми клетками, расположенными вокруг места поражения, меристематической активности. Возникает перидерма, отгораживающая очаг инфекции. Судьба же клеточной стенки, атрибута растительной клетки, неоднозначна. Существуют два возможных пути ее преобразования, диктуемые привязкой к событию и различающиеся конечным результатом.
Во-первых, клеточная стенка может упрочняться благодаря сшивке белков, образованию целлюлозных утолщений и лигнификации. Укрепление клеточной стенки затрудняет проникновение патогена в клетку или, напротив, способствует замуровыванию уже проникшего микроба внутри клетки. При формировании жестких сосудов проводящей системы (трахеидов ксилемы и ситовидных элементов флоэмы) на стенках соответствующих клеток образуются утолщения, которые затем укрепляются отложением лигнина.
Второй путь – разрушение клеточной стенки при участии активирующихся гидролитических ферментов. Тотальное разрушение клеточных стенок происходит при аэренхимогенезе [15]. Гидролазы действуют локально и частично растворяют клеточную стенку в отделительном слое при листопаде и при отделении созревших плодов [22].
У растений, как и у животных, наряду с апоптозом, существует некроз. Так, Н2О2 в малых концентрациях – индуктор апоптоза, в высоких концентрациях вызывает быструю гибель клеток, без каких-либо морфологических изменений, характерных для апоптоза [24].
Итак, наряду с делением, дифференцировкой, клеточная смерть является процессом, способствующим нормальному становлению и функционированию организма, необходимым и благоприятным для организма в целом.
Молекулярные механизмы апоптоза
Апоптоз – многоэтапный процесс. Первый этап – прием сигнала, предвестника гибели в виде информации, поступающей к клетке извне или возникающей в недрах самой клетки. Сигнал воспринимается рецептором и подвергается анализу. Далее через рецепторы или их сочетания полученный сигнал последовательно передается молекулам-посредникам (мессенджерам) различного порядка и в конечном итоге достигаетядра, где и происходит включение программы клеточного самоубийства путем активации летальных и/или репрессии антилетальных генов. Однако существование ПКС в безъядерных системах (цитопластах – клетках, лишенных ядра) показывает, что наличие ядра не является обязательным для реализации процесса [32].
Применительно к клеткам животных и человека апоптоз в большинстве случаев связан с протеолитической активацией каскада каспаз – семейства эволюционно консервативных цистеиновых протеаз, которые специфически расщепляют белки после остатков аспарагиновой кислоты (см. обзоры [33–35]). На основе структурной гомологии каспазы подразделяются на подсемейства а) каспазы-1 (каспазы 1, 4, 5), б) каспазы-2 (каспаза-2) и в) каспазы-3 (каспазы 3, 6–10) [36]. Цистеиновые протеазы, по-видимому, участвуют также в ПКС у растений [37]. Однако апоптоз возможен и без участия каспаз: сверхсинтез белков-промоторов апоптоза Bax и Bak индуцирует ПКС в присутствии ингибиторов каспаз [38, 39].
В результате действия каспаз происходит [33–36]:
активация прокаспаз с образованием каспаз;
расщепление антиапоптозных белков семейства Bcl-2. Подвергается протеолизу ингибитор ДНКазы, ответственный за фрагментацию ДНК. В нормальных клетках апоптозная ДНКаза CAD (caspase-activated DNase) образует неактивный комплекс с ингибитором CAD, обозначаемым ICAD или DFF (DNA fragmentation factor). При апоптозе ингибитор ICAD с участием каспаз 3 или 7 инактивируется [40], и свободная CAD, вызывая межнуклеосомальные разрывы хроматина, ведет к образованию фрагментов ДНК с молекулярной массой, кратной молекулярной массе ДНК в нуклеосомных частицах – 180-200 пар нуклеотидов. Эти фрагменты при электрофоретическом разделении в агарозном геле дают характерную "лесенку ДНК". Апоптоз возможен и без фрагментации ДНК [2]. Обнаружен ядерный белок Acinus (apoptotic chromatin condensation inducer in the nucleus), из которого при комбинированном действии каспазы-3 (протеолиз при Asp 1093) и неидентифицированной протеазы (протеолиз при Ser 987) образуется фрагмент Ser 987 – Asp 1093. Этот фрагмент в присутствии дополнительных неядерных факторов вызывает апоптотическую конденсацию хроматина и фрагментацию ядра (кариорексис) без фрагментации ДНК [41, 42];
гидролиз белков ламинов, армирующих ядерную мембрану. Это ведет к конденсации хроматина;
разрушение белков, участвующих в регуляции цитоскелета;
инактивация и нарушение регуляции белков, участвующих в репарации ДНК, сплайсинге мРНК, репликации ДНК. Мишенью каспаз является поли(ADP-рибозо)полимераза (ПАРП). Этот фермент участвует в репарации ДНК, катализируя поли(ADP-рибозилирование) белков, связанных с ДНК (см. обзоры [3,11]). Донором ADP-рибозы является NAD+. Активность ПАРП возрастает в 500 раз и более при связывании с участками разрыва ДНК. Апоптотическая гибель клетки сопровождается расщеплением ПАРП каспазами. Чрезмерная активация ПАРП при массированных разрывах ДНК, сильно снижая содержание внутриклеточного NAD+, ведет к подавлению гликолиза и митохондриального дыхания и вызывает гибель клетки по варианту некроза.
Существует несколько путей реализации программы ПКС [2, 34, 43–46].
1. Среди них важное место занимает путь, опосредованный физиологическими индукторами, действие которых реализуется через клеточные рецепторы [34, 47–52], специально предназначенные для включения программы апоптоза. Этот путь передачи сигнала ПКС схематически можно изобразить следующим образом: индукторы ’ рецепторы ’ адаптеры ’ каспазы первого эшелона ’ регуляторы ’ каспазы второго эшелона. Так, рецептор, обозначаемый Fas, взаимодействуя с соответствующим лигандом (лигандом FasL), трансмембранным белком Т-киллера, активируется и запускает программу смерти клетки, инфицированной вирусом. Тем же путем при взаимодействии с лигандом FasL на поверхности ТН-1-лимфоцитов или с антителом к Fas-рецептору погибают ставшие ненужными выздоровевшему организму В-лимфоциты, продуценты антител, несущие Fas-рецептор. FasL– лиганд, относящийся к многочисленному семейству фактора некроза опухолей (TNF – tumor necrosis factor). Это семейство гомотримерных лигандов, кроме FasL и TNFa , включает TNFb (лимфотоксин), TRAIL (Apo2L), CD40L, CD27L, CD30L, OX40L.
Fas – член семейства рецепторов TNF. Все они представлены трансмембранными белками, которые внеклеточными участками взаимодействуют с тримерами лигандов-индукторов (рис. 2). Взаимодействие рецептора и лиганда приводит к образованию кластеров рецепторных молекул и связыванию их внутриклеточных участков с адаптерами. Адаптер, связавшись с рецептором, вступает во взаимодействие с эффекторами, пока еще неактивными предшественниками протеаз из семейства каспаз первого эшелона (инициирующих каспаз).
Взаимодействие адаптера с рецептором и эффектором осуществляется через гомофильные белок-белковые взаимодействия небольших доменов: DD (death domain – домен смерти), DED (death-effector domain – домен эффектора смерти), CARD (caspase activation and recruitment domain – домен активации и рекрутирования каспазы). Все они имеют сходную структуру, содержат по шесть a-спиральных участков [45, 46]. Домены DD участвуют во взаимодействии рецептора Fas c адаптером FADD (Fas-associated DD-protein) и во взаимодействии рецепторов TNFR1 и DR3 (death receptor 3) с адаптером TRADD (TNFR1-associated DD-protein). Домены DED участвуют во взаимодействии адаптера FADD с прокаспазами 8 и 10. Адаптер RAIDD (RIP-associated Ich-1/CED-3 homologous protein with a death domain, RIP – receptor interacting protein) связывается с прокаспазой-2 через CARD-домены [ 45, 46, 51].
Наиболее подробно охарактеризована прокаспаза-8 (FLICE/MACH/Mch5), рекрутируемая рецептором Fas через адаптeр FADD. Образуются агрегаты FasL – Fas – FADD – прокаспаза-8. Подобные агрегаты, в которых происходит активация каспаз, названы апоптосомами [43], апоптозными шаперонами [53], или сигнальными комплексами, индуцирующими смерть (DISC – death-inducing signaling complex) [49].
Прокаспазы обладают незначительной протеолитической активностью, составляющей 1–2% активности зрелой каспазы [34, 43, 54]. Будучи в мономерной форме, прокаспазы, концентрация которых в клетке ничтожна, находятся в латентном состоянии. Предполагается, что пространственное сближение молекул прокaспаз при их агрегации ведет к образованию активных каспаз через механизм протеолитического само- и перекрестного расщепления (ауто- или транс-процессинга) [34, 43, 54]. В результате от прокаспазы (молекулярная масса 30–50 кДа) отделяется регуляторный N-концевой домен (продомен), а оставшаяся часть молекулы разделяется на большую (~20 кДа) и малую (~10 кДа) субъединицы (рис. 3). Затем происходит ассоциация большой и малой субъединиц. Два гетеродимера образуют тетрамер с двумя каталитическими участками, действующими независимо друг от друга. Таким образом прокаспаза-8 активируется и высвобождается в цитоплазму в виде каспазы-8.
Существуют другие пути активации каспазы-8 – с участием рецепторов TNFR1 и DR3 [51]. Однако эти пути, включаемые одним и тем же адаптером TRADD, конкурируют с параллельными путями активации ядерных факторов транскрипции NF-єB (nuclear factor kappa B) и JNK/AP-1 (JNK, Jun-N-концевая киназа, является компонентом митоген-активируемого киназного пути, ведущего к активации фактора транскрипции AP-1), зависимыми от адаптеров RIP и TRAF (TNFR1-associated factor).под контролем этих факторов транскрипции находится синтез белковых регуляторов, которые блокируют TNF- или Apo3L-индуцированную активацию каспазы-8. Предполагаются следующие пути передачи про- и антиапоптозных сигналов [51]:
На этапе активации каспаз первого эшелона жизнь клетки еще можно сохранить. Существуют регуляторы, которые блокируют или, напротив, усиливают разрушительное действие каспаз первого эшелона (см. обзоры [36, 52, 55–58]). К ним относятся белки Bcl-2 (ингибиторы апоптоза: A1, Bcl-2, Bcl-W, Bcl-XL, Brag-1, Mcl-1 и NR13) и Bax (промоторы апоптоза: Bad, Bak, Bax, Bcl-XS, Bid, Bik, Bim, Hrk, Mtd). Эти белки эволюционно консервативны: гомолог Bcl-2 обнаружен даже у губок Geodia cydomium и Suberites domuncula, у которых апоптоз необходим для морфогенеза [59].
Каспаза-8 активирует каспазу второго эшелона (эффекторную каспазу): путем протеолиза из прокаспазы-3 образуется каспаза-3, после чего процесс, запущенный программой смерти, оказывается необратимым. Рис. 4 иллюстрирует взаимодействие между каспазами первого и второго эшелона. Каспаза-3 способна в дальнейшем к самостоятельной активации (автокатализу или автопроцессингу), активирует ряд других протеаз семейства каспаз, активирует фактор фрагментации ДНК, ведет к необратимому распаду ДНК на нуклеосомальные фрагменты. Так запускается каскад протеолитических ферментов,осуществляющих апоптоз.
2. В клетках, подвергшихся воздействию индуктора апоптоза, резко снижается мембранный потенциал (Dy)митохондрий [34, 55, 58, 60, 61]. Падение Dy обусловлено увеличением проницаемости внутренней мембраны митохондрий (permeability transition) вследствие образования гигантских пор [62]. Разнообразны факторы, вызывающие раскрытие пор [60]. К ним относятся истощение клеток восстановленным глутатионом, NAD(P)H, ATP и ADP, образование активных форм кислорода, разобщение окислительного фосфорелирования протонофорными соединениями, увеличение содержания Ca2+ в цитоплазме. Образование пор в митохондриях можно вызвать церамидом, NO, каспазами, амфипатическими пептидами, жирными кислотами [60]. Поры имеют диаметр 2,9 нм, позволяющий пересекать мембрану веществам с молекулярной массой 1,5 кДа и ниже. Следствием раскрытия поры является набухание митохондриального матрикса, разрыв наружной мембраны митохондрий и высвобождение растворимых белков межмембранного объема [63]. Среди этих белков – ряд апоптогенных факторов: цитохром с [64–66], прокаспазы 2, 3 и 9 [67, 68], белок AIF (apoptosis inducing factor), представляющий собой флавопротеин с молекулярной массой 57 кДа [69]. Прокаспаза-3 обнаруживается как в межмембранном объеме митохондрий, так и в цитоплазме [67].
Образование гигантских пор не является единственным механизмом выхода межмембранных белков митохондрий в цитоплазму. Предполагается [61], что разрыв наружной мембраны митохондрий может быть вызван гиперполяризацией внутренней мембраны (ср. с гипополяризацией при раскрытии гигантских пор). Возможен и альтернативный механизм, без разрыва мембраны, – раскрытие гигантского белкового канала в самой наружной мембране, способного пропускать цитохром с и другие белки из межмембранного пространства [61].
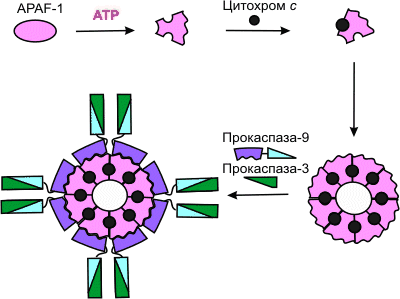
Высвобождаемый из митохондрий цитохром с вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) участвует в активации каспазы-9 [70]. APAF-1 – белок с молекулярной массой 130 кДа, содержащий CARD-домен (caspase activation and recruitment domain) на N-конце и 12 повторяющихся аминокислотных WD-40-последовательностей (WD – дипептид из триптофана и аспартата) на С-конце [71], образует комплекс с прокаспазой-9 в присутствии цитохрома с и dATP или АТР [70] (концентрация dATP в клетке в 1000 раз ниже концентрации АТР [72]). К наиболее охарактеризованным WD-белкам относится b -cубъединица G-белков. Из этих субъединиц собираются жесткие, симметричные структуры, наподобие веера или пропеллера (см. обзор [73]). WD-Повторы свойственны белкам, участвующим в регуляции деления и дифференцировки эукариотных клеток, транскрипции генов, модификации мРНК, трансмембранной передачи сигналов, слияния мембранных везикул. Среди прокариот WD-белки обнаружены у цианобактерий [74, 75].
APAF-1 играет роль арматуры, на которой происходит аутокаталитический процессинг каспазы-9 [76–78]. Предполагается, что в результате зависимого от гидролиза dATP (или АТР) конформационного изменения APAF-1 приобретает способность связывать цитохром с (рис. 5). Связав цитохром с, APAF-1 претерпевает дальнейшее конформационное изменение, способствующее его олигомеризации и открывающее доступ CARD-домена APAF-1 для прокаспазы-9, которая тоже содержит CARD-домен. Так образуется конструкция, называемая тоже апоптосомой, с молекулярной массой > 1,3 млн дальтон, в составе которой – не менее 8 субъединиц APAF-1 [76]. Благодаря гомофильному CARD-CARD-взаимодействию с APAF-1 в эквимолярном соотношении связывается прокаспаза-9, а затем прокаспаза-9 связывает прокаспазу-3. Пространственное сближение молекул прокаспазы-9 на мультимерной арматуре из APAF-1-цитохром-с-комплексов, по-видимому, приводит к межмолекулярному протеолитическому процессингу прокаспазы-9 с образованием активной каспазы-9 [76-78]. Сходный механизм предложен для активации прокаспазы CED-3 у червя Caenorhabditis elegans [79] –аналога прокаспазы-9 млекопитающих. Альтернативный вариант – прокаспаза-9, связавшись с апоптосомой, может принять конформацию, которая приводит к внутримолекулярному процессингу (самоактивации) [77]. Зрелая каспаза-9 затем расщепляет и активирует прокаспазу-3. Мутантный APAF-1, лишенный WD-40-повторов, активирует прокаспазу-9, но не способен к рекрутированию и активации прокаспазы-3 [77].
Флавопротеин AIF, будучи добавленным к изолированным ядрам из клеток HeLa, вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям печени крыс – высвобождение цитохрома с и каспазы-9 [69]. Микроинъекция AIF в интактные фибробласты крыс приводит к конденсации хроматина по переферии ядра, разрыву ДНК на крупные фрагменты длиной 50 т.п.н. и больше, диссипации Dy в митохондриях и переходу фосфатидилсерина из внутреннего слоя цитоплазматической мембраны в наружный. Ни один из этих эффектов AIF не предотвращается пептидным ингибитором каспаз N-бензоилоксикарбонил-Val-Ala-Asp.трифторметилкетоном (Z-VAD.fmk), который предотвращает апоптоз, индуцированный микроинъецированным цитохромом с. Эти данные показывают, что AIF является митохондриальным эффектором ПКС у животных, действующим независимо от каспаз [69].
Кроме рассмотренных компонентов, при нарушении наружной мембраны митохондрий из межмембранного объема выделяется термолабильный фактор, вызывающий необратимое превращение ксантиндегидрогеназы в ксантиноксидазу [80]. Фактор устойчив к ряду испытанных ингибиторов протеаз, включая каспазы, сериновые и металлопротеазы. Ксантиндегидрогеназа катализирует зависимое от NAD+ окисление ксантина до гипоксантина и последующее окисление гипоксантина до мочевой кислоты. Ксантиноксидаза катализирует те же реакции, но не с NAD+, а с О2 в качестве акцептора электронов. При этом образуются О2A, Н2О2, а из них – и другие активные формы кислорода (АФК), которые разрушают митохондрии и являются мощными индукторами апоптоза. Механизмы образования АФК, конечно, не ограничиваются ксантиноксидазной реакцией. Главным источником АФК в клетках являются митохондрии. Резкое увеличение АФК происходит при возрастании мембранного потенциала в митохондриях, когда снижено потребление ATP и скорость дыхания лимитируетсяADP [81]. Доля электронного потока через дыхательную цепь митохондрий, идущая на образование О2A, достигает 1-5 % (см. [61]). Цитоплазматическая мембрана макрофагов и нейтрофилов, как уже отмечалось, содержит О2A – генерирующую NADPH-оксидазу.
В зависимости от пути, по которому осуществляется активация каспаз, различают разные типы клеток [82]. Клетки типа I (в частности, линия лимфобластоидных В-клеток SKW и T-клетки линии Н9) подвергаются ПКС по пути, зависимому от апоптозных рецепторов плазматической мембраны без участия митохондриальных белков. Клетки типа II (например, линии Т-клеток Jurkat и СЕМ) погибают по пути апоптоза, зависимому от митохондриального цитохрома с [82]. ПКС, вызванная химиотерапевтическими соединениями, УФ- или і-облучением, по-видимому, напрямую связана с апоптозной функцией митохондрий: клетки, лишенные генов белка APAF-1 или каспазы-9, устойчивы к химио- и радиационной обработке, но погибают при индукции Fas-рецептора [83–86].
Некоторые клетки, например, клетки эмбриональной нервной системы, включают механизмы апоптоза, если они испытывают дефицит апоптозподавляющих сигналов (называемых также факторами выживания) от других клеток. Физиологический смысл процесса – в элиминации избыточных нервных клеток, конкурирующих за ограниченный фонд факторов выживания. Эпителиальные клетки при отделении от внеклеточного матрикса, вырабатывающего факторы выживания, тоже обречены на ПКС. Факторы выживания связываются соответствующими цитоплазматическими рецепторами, активируя синтез подавляющих апоптоз агентов и блокируя стимуляторы апоптоза [44]. Некоторые вещества (например, стероидные гормоны) оказывают дифференцированный эффект на различные типы клеток – предотвращают апоптоз одних типов клеток и индуцируют его у других [2].
Так, при наличии во внеклеточном матриксе факторов роста PDGF (platelet-derived growth factor – тромбоцитарный фактор роста) или NGF (nerve growth factor – фактор роста нервов) и цитокина интерлейкина-3 (IL-3) проапоптозный белок Bad не активен (см. обзор [58]). Факторы роста, связавшись со своим рецептором на плазматической мембране, вызывают активацию цитозольной протеинкиназы В, обозначаемой Akt/PKB/RAC и катализирующей фосфорилирование Bad по Ser-136. IL-3 тоже связывается со своим рецептором на плазматической мембране и активирует митохондриальную cAMP-зависимую протеинкиназу А (РКА), катализирующую фосфорилирование Bad по Ser-112. Будучи фосфорилированным по обоим остаткам серина, Bad образует комплекс с белком 14-3-3, располагающийся в цитоплазме. Дефицит факторов роста и IL-3 воспринимается клеткой как сигнал к апоптозу: происходит дефосфорилирование Bad, его внедрение в наружную мембрану митохондрий, выход цитохрома с из митохондрий и последующая активация каспазы-9 через APAF-1-зависимый механизм. Кроме этого дефицит IL-3 вызывает перемещение мономерного проапоптозного белка Bax из цитоплазмы в наружную мембрану митохондрий, последующая сшивка молекул Bax с образованием гомодимеров тоже ведет к выходуцитохрома с из митохондрий и гибели клетки.
3. В ряде случаев ПКС реализуется в результате комбинированного действия двух путей – с участием и рецепторов плазматической мембраны, и митохондриального цитохрома с. Так, повреждение ДНК ведет к накоплению в клетке белкового продукта гена р53, который может останавливать деление клеток и/или индуцировать апоптоз (см. обзоры [87–89]). У более чем 50% изученных видов опухолевых клеток ген р53 инактивирован [44], у них нарушена р53-зависимая регуляция клеточного гомеостаза.
Белок р53 является фактором транскрипции, регулирующим активность ряда генов. Предполагается, что ответная реакция на образование белка р53 зависит от степени нарушения клеточного генома [89]. При умеренном нарушении генома происходит остановка клеточного деления, осуществляется репарация ДНК, и клетка продолжает свое существование. При чрезмерном нарушении генома, когда ДНК уже не поддается репарации, включаются рецепторный и цитохром с-зависимый апоптозные каскады активации каспаз.
Различные пути апоптоза могут взаимодействовать между собой. В некоторых случаях зависимый от рецепторов путь ведет к малоэффективной активации прокаспазы-8. В этом случае подключается зависимый от митохондрий путь апоптоза: каспаза-8 (образовавшаяся в небольших количествах) взаимодействует в цитоплазме с белком Bid из семейства Bax, расщепляя его надвое. С-Концевой домен Bid далее внедряется в митохондриальную мембрану, индуцируя выход цитохрома с из митохондрий и его связывание с APAF-1 [43, 58].
4. Существует путь передачи сигнала ПКС с участием эндоплазматического ретикулума (ЭР) [90, 91]. В ЭР локализована прокаспаза-12. Нарушение внутриклеточного Ca2+-гомеостаза добавкой тапсигаргина или Ca2+-ионофорного антибиотика А23187 ведет к апоптозу клеток, вызванному превращением прокаспазы-12 в каспазу-12. ЭР-зависимый апоптоз связан с болезнью Альцгеймера: кортикальные нейроны мышей, дефицитных по каспазе-12, устойчивы к апоптозу, индуцированному І-амилоидным белком, но не к апоптозу с участием рецепторов плазматической мембраны или митохондриального цитохрома с.
5. Цитотоксические лимфоциты, Т-киллеры, могут вызывать апоптоз у инфицированных клеток с помощью белка перфорина. Полимеризуясь, перфорин образует в цитоплазматической мембране клетки-мишени трансмембранные каналы, по которым внутрь клетки поступают TNFb , гранзимы (фрагментины) – смесь сериновых протеаз. Существенным компонентом этой смеси является гранзим В – протеолитический фермент, превращающий прокаспазу-3 в активную каспазу-3 [2, 43].
6. Взаимодействие клеток с внеклеточным матриксом осуществляется с помощью интегринов. Интегрины – большое семейство гетеродимерных мембранных белков, которые участвуют в адгезии клеток, связывая внутриклеточный цитоскелет с лигандами внеклеточного матрикса. Нарушение адгезии клеток индуцирует апоптоз. Большинство интегринов специфическивзаимодействует с трипептидным RGD (аргинин-глицин-аспартат)-мотивом, входящим в состав белков внеклеточного матрикса. Растворимые низкомолекулярные RGD-содержащие пептиды являются эффективными индукторами апоптоза: проникая в клетки, они активируют латентную каспазу-3 [92, 93]. Ряд каспаз, включая каспазу-3, содержит RGD-последовательность вблизи активного центра фермента. В молекуле прокаспазы эта последовательность, вероятно, вовлечена во внутримолекулярное взаимодействие, придающее молекуле профермента такую конформацию, при которой протеазная активность не может проявиться. Предположительно RGD-последовательность взаимодействует с последовательностью DDM (аспартат-аспартат-метионин), локализованной вблизи участка протеолитической активации прокаспазы-3. Низкомолекулярный RGD-пептид, проникая в клетку и вступая в конкурентные взаимоотношения с RGD-последовательностью прокаспазы-3, вытесняет ее из сферы взаимодействия с DDM-последовательностью молекул профермента и индуцирует изменение их конформации, олигомеризацию и аутопроцессинг прокаспазы-3 с образованием активной каспазы-3 [92].
7. Особую форму апоптоза претерпевают эритроциты млекопитающих. Биогенез эритроцитов из плюрипотентной стволовой клетки в костном мозге включает ряд промежуточных этапов. На этапе эритробласта ядро изгоняется (выталкивается) из клетки и пожирается макрофагом [94, 95]. Альтернативный вариант: кариорексис (деструкция ядра) с образованием телец Жолли и их последующий распад и лизис внутри клетки [94]. Безъядерная клетка, называемая ретикулоцитом, в дальнейшем теряет митохондрии и рибосомы и превращается в эритроцит. Потерю ядра эритробластом можно рассматривать как особую форму ядерного апоптоза. Выяснение его механизма позволило бы применить его для обезвреживания опухолевых клеток. Эритроцит человека функционирует около 4 месяцев, а затем, поизносившись, исчезает в недрах ретикулоэндотелиальной системы, не причиняя неудобств окружающим клеткам. Лишенный ядра и митохондрий эритроцит, исполнив свое назначение, по-видимому, включает программу гибели, чтобы после этого поступить в распоряжение макрофагов печени и селезенки. Однако ингибитор протеинкиназы стауроспорин и ингибитор синтеза белка циклогексимид (индуцирующий ПКС у большинства испытанных типов клеток млекопитающих) не вызывает ПКС у безъядерных эритроцитов человека [96]. Стауроспорин и циклогексимид, а также отсутствие сыворотки в среде инкубации индуцируют гибель эритроцитов цыпленка (содержащих транскрипционно неактивное клеточное ядро) с выраженными признаками апоптоза по пути, который реализуется без участия каспаз. Сперматозоиды мыши, у которых ядра тоже не обладают активностью в транскрипции ДНК, при инкубации в искусственных средах спонтанно погибают за 1–2 суток; стауроспорин, циклогексимид и пептидный ингибтор каспаз z-VAD.fmk не ускоряют и не замедляют клеточную гибель [97].
Мало известно о механизме ПКС у растений. В сравнении с естественными индукторами ПКС химические и физические воздействия методически более привлекательны, поскольку вызывают синхронный апоптоз с высоким выходом погибших клеток, что облегчает последующий анализ результатов. Так, апоптоз у растений можно вызвать обработкой CN– [98-100], менадионом [101], тепловым воздействием [102].
Показано [100], что NaCN вызывает разрушение ядер в эпидермальных и устьичных клетках листьев гороха. Устьичные клетки значительно устойчивее к CN–, чем эпидермальные. Свет ускоряет CN.-индуцированноеразрушение ядер в устьичных клетках. Эффект света незначителен на эпидермальных клетках, которые, в отличие от устьичных клеток, не содержат хлоропластов. Эти данные могут указывать на возможное участие хлоропластов в CN–-индуцированной гибели устьичных клеток. Антиоксиданты (ионол и витамин Е) тормозят CN–-индуцированное разрушение ядер в эпидермальных клетках. Витамин Е в значительной степени снимает эффект CN– на устьичные клетки. Салициловая кислота – ингибитор каталазы и аскорбатпероксидазы – индуцирует 100%-ное разрушение ядер в эпидермальных клетках, но не оказывает значительного влияния на состояние ядерного аппарата в устьичных клетках. Предполагается, что CN–, ингибируя каталазу и пероксидазы, приводит к образованию и накоплению АФК, индуцирующих апоптоз. Подобно митохондриям, играющим важную роль в апоптозе животных, возможно участие хлоропластов в апоптозе растений [100].
Гиперчувствительный ответ на заражение патогенными возбудителями тоже сопровождается накоплением АФК в клетках растений. Это обусловлено подавлением экспрессии аскорбатпероксидазы и каталазы. Трансгенные растения табака, у которых синтез этих ферментов подавлен, гиперчувствительны к патогенам: у них ПКС вызывается низкими дозами патогенов, которые не оказывают влияния на контрольные растения [103].
Действие менадиона как индуктора апоптоза, по-видимому, тоже связано с образованием АФК: восстанавливаясь компонентами дыхательной цепи митохондрий, менадион спонтанно окисляется О2 в одноэлектронной реакции. Обработка протопластов табака менадионом ведет к выходу цитохрома с из митохондрий в цитоплазму, деградации поли(ADP-рибозо)полимеразы (ПАРП), фрагментации ДНК [101]. Тетрапептидные ингибиторы каспаз предотвращают расщепление ПАРП (субстрата каспазы-3 у животных), но не влияют на выход цитохрома с из митохондрий. Выход цитохрома с в цитоплазму и фрагментация ДНК наблюдаются также при тепловой обработке (55Ъ, 10 мин) семядолей этиолированных проростков огурца [102].
Экспрессия проапоптозного белка Bax вызывает ПКС у растений табака [104]. Bax-индуцированная ПКС фенотипически сходна с гибелью клеток при гиперчувствительном ответе на заражение вирусом табачной мозаики. В рисе и арабидопсисе выявлен ген ингибитора белка Bax. Экспрессия этого гена подавляет Bax-индуцированную гибель дрожжей Saccharomyces cerevisiae [105].
Таким образом, имеющиеся данные свидетельствуют об общности механизмов ПКС у животных и растений.
Программируемая гибель у микроорганизмов
По данным анализа полностью и частично расшифрованных геномов, у микроорганизмов идентифицированы белки, соответствующие апоптозным факторам млекопитающих [46]. Так, у слизистого гриба из группы акразиевых, Dictyostelium discoideum содержится адаптер TRAF. У дрожжей обнаружен лиганд LRR (leucine-rich repeat), адаптерные домены BIR и MATH, у бактерий – лиганд LRR, адаптерный домен TIR, АТРазный домен фактора APAF-1, гомолог каспазы. Эти микробные домены, по-видимому, вовлечены в различные регуляторные механизмы [46].
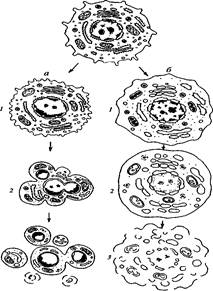
Гриб D. discoideum имеет сложный цикл развития (рис. 6). В этом цикле фаза обособленных амебоидных вегетативных клеток, размножающихся бинарным делением, сочетается с формированием видимых невооруженным глазом подвижных клеточных скоплений (мигрирующего псевдоплазмодия) и далее с построением многоклеточного плодового тела, в котором отдельные амебы покрываются оболочкой (инцистируются) и превращаются в споры, прорастающие в благоприятных условиях в амебоидные вегетативные клетки. Образование псевдоплазмодия – реакция на нехватку пищи (см. обзоры [106–108]). Клетки на переднем конце псевдоплазмодия погибают с признаками ПКС [109]. Из погибших клеток формируется ножка плодового тела. Процесс находится под влиянием ряда межклеточных коммуникационных агентов: сАМР и в особенности 1-(3,5-дихлор-2,6-гидрокси-4-метоксифенил)-1-гексанона [107]. Ингибиторы каспаз не предотвращают гибели клеток в ножке плодового тела D. discoideum , т.е. ПКС в данной системе, по-видимому, не зависит от классических механизмов апоптоза [110].
Процесс, аналогичный ПКС, у ресничных инфузорий – разрушение макронуклеуса (большего клеточного ядра) с деградацией его ДНК, наступающее в связи с конъюгацией, необходимой для рекомбинации ДНК в малом клеточном ядре – микронуклеусе [111].
Паразитический жгутиконосец Trypanosoma cruzi имеет три основные стадии жизненного цикла: эпимастиготы, трипомастиготы и амастиготы. Эпимастиготы интенсивно делятся, обитая в кишечнике кровососущих насекомых. Они далее дифференцируются в неделящихся трипомастигот, которые, попадая на кожу позвоночного животного (или человека) с экскрементами кровососущих насекомых, внедряются в клетки позвоночных и превращаются в активно делящихся внутриклеточных паразитов амастигот. Амастиготы продуцируют новые трипомастиготы, которые выходят из клеток и распространяются с током крови, попадая в новые клетки (и давая амастиготы) или в кишечник насекомого-кровососа (превращаясь в эпимастиготы). Установлено, чтопри выращивании T. cruzi in vitro при 27оС (моделирование условий кишечника насекомого) эпимастиготы через некоторое время образуют трипомастиготы, причем появление трипомастигот сопряжено с массовой (программируемой?) гибелью эпимастигот. Предполагаемый смысл ПКС – неделящиеся трипомастиготы, необходимые для распространения паразита на позвоночного хозяина, следует оберегать от конкуренции за ресурсы с интенсивно пролиферирующими эпимастиготами [112].
Рассмотренные формы клеточной гибели у Protozoa связаны с процессами дифференцировки и проявляют функциональную общность с ПКС при онтогенетическом развитии животных и растений, например, при отмирании хвоста у головастика и формировании листьев у растений.
Дрожжи не содержат регуляторных факторов апоптоза, подобных Bcl-2 и Bax. Такие белки отсутствуют и у растений [25]. Однако рост Saccharomyces сerevisiae и Schizosaccharomyces pombe подавляется при экспрессии проапоптозных белков Baх и Bak. Дрожжевые клетки погибают с характерной для апоптоза картиной (накопление фосфатидилсерина во внешнем монослое цитоплазматической мембраны, конденсация хроматина, фрагментация ДНК, распад клеток на апоптозные везикулы). ПКС дрожжей предотвращается при соэкспрессии антиапоптозных белков Bcl-2 или Bcl-XL. [57, 113, 114]. Cверхэкспрессия Bcl-XL у растений табака не блокирует ПКС в ответ на вирусную и бактериальную инфекции [115]. Поскольку гибель Вах-экспрессирующих клеток у бродящих S. cerevisiae (отсутствуют митохондрии) выражена слабее, чем у дышащих (имеются митохондрии), предполагается, что цитохром с и митохондриальный переносчик адениннуклеотидов участвуют в Вах-индуцированном ПКС дрожжей [116].
Проапоптозные белки активны у дрожжей, хотя их клетки не синтезируют каспазы. Экспрессия каспазы-3 тормозит рост S. сerevisiae, но не вызывает их гибели [117]. Торможение снимается при нарушении структуры активного центра каспазы-3 в результате мутации. Соэкспрессия вирусного ингибитора каспазы-3 или добавление пептидного ингибитора z-VAD.fmk снижают действие экспрессированной каспазы-3 на рост клеток [117]. У животных Bax индуцирует ПКС в присутствии ингибиторов каспаз. Этот путь ПКС предположительно связан с накоплением в клетке активных форм кислорода. Вероятно, существует общий для дрожжей и млекопитающих эволюционно-консервативный бескаспазный путь ПКС, зависящий от активных форм кислорода [118].
Описанные примеры ПКС у дрожжей связаны с экспрессией чужеродных генов, не имеющих аналогов в геноме дрожжей. Однако ПКС характерна для S. сerevisiae и в ходе реализации программы нормального развития. Так, материнская клетка, от которой отпочковываются дочерние клетки S. сerevisiae, по-видимому, элиминируется после формирования на ней 20-30 почек [2].
Что ожидать от прокариот, если у них экспрессировать белки, замешанные в механизме ПКС у млекопитающих? Такие эксперименты проведены на Escherichia coli [119]: клетки бактерии погибают при экспрессии внутриклеточной части рецептора Fas T-лимфоцитов человека.
Прокариотическим аналогом апоптоза можно считать гибель части клеточной популяции E. coli и ряда других бактерий в условиях стазиса – остановки роста бактериальной популяции (при исчерпании питательного субстрата, под влиянием того или иного стрессорного фактора). Так, голодающая популяция E. coli разделяется на две субпопуляции, одна из которых гибнет и подвергается автолизу, в то время как другая субпопуляция использует продукты автолиза как питательный субстрат и продолжает расти, синтезировать РНК (судя по включению 3Н-уридиновой метки) и формировать колониеобразующие единицы [120]. Раскрыт генетическиймеханизм ПКС в подобных прокариотических системах [121, 122]. Геном E. coli содержит оперон с двумя генами: mazE и mazF (рис. 7). Ген mazF кодирует стабильный цитотоксический белок, а mazE – нестабильное противоядие к белку MazF, быстро разрушаемое протеазой. В условиях голодания, когда происходит исчерпание фонда свободных аминокислот активируется оперон rel, чей белковый продукт RelA отвечает за синтез гуанозинтетрафосфата. Гуанозинтетрафосфат блокирует экспрессию обоих генов: противоядие разрушается, и в результате стабильный белок-яд MazF вызывает гибель и автолиз части популяции, тем самым пополняя фонд аминокислот и вновь активируя синтез противоядия MazF у оставшихся в живых клеток E. coli [121, 122].
Система mazE-mazF, расположенная на хромосоме E. coli, аналогична многочисленным плазмидным системам, которые кодируют стабильный цитотоксический агент в комбинации с лабильным противоядием к нему (такие плазмидные системы именуются модулями зависимости, addiction modules). Так, у E. coli обнаружена плазмида, содержащаяген стабильной ДНК-рестриктазы и нестабильной ДНК-метилазы, которая предохраняет ДНК от рестриктазы (метилированная ДНК не может быть узнана рестриктазой). Утрата плазмиды влечет за собой прекращение синтеза метилазы, и стабильная рестриктаза фрагментирует вновь снтезируемую неметилированную ДНК и вызывает гибель потерявших плазмиду клеток [123]. Поэтому подобные модули зависимости рассматривают как "эгоистичные ДНК" (термин Р. Докинза [124]): гибнут те клетки, которые пытаются избавиться от этой ДНК; сохраняются те, которые распространяют ее.
Участки hok/sok и pnd плазмид R1 и R483 [125] кодируют токсины, вызывающие диссипацию мембранного потенциала и наряду с этим гены, транскрипция которых дает лабильные антисмысловые РНК, препятствующие транскрипции плазмидной ДНК. Плазмида F y E. coli несет гены, отвечающие за синтез 1)токсина CcdB, который превращает ДНК-гиразу в ДНК-повреждающий агент и 2)белка-антидота CcdA, образующего с CcdB неактивный комплекс [123]. Приведенные примеры относятся к внутриклеточным механизмам ПКС, элиминирующим ту самую клетку, которая утратила плазмиду с модулем зависимости.
Имеются и внеклеточные механизмы ПКС: содержащие плазмиду клетки убивают соседей, не имеющих этой плазмиды. Речь идет о бактерицинах и подобных им агентах, чьи гены расположены на плазмидах вместе с генами, определяющими устойчивость к токсину самой бактериоцин-продуцирующей клетки. Внеклеточные токсины используют те же механизмы элиминирования чувствительных клеток, что и рассмотренные выше внутриклеточные токсины. Так, колицин Е1 и сходные с ним агенты, подобно плазмидам R1 и R483, ведут к деэнергизации цитоплазматической мембраны E. coli, а микроцин В17 (как и белок CcdB, кодируемый плазмидой F) трансформирует ДНК-гиразу в ДНК-повреждающий агент [123]. Известны также колицины (Е9), разрушающие ДНК; расщепляющие рибосомальную РНК и поэтому препятствующие синтезу белка; ингибирующие синтез пептидогликана клеточной стенки [126]. Колициногенные клетки E. coli защищаются от действия колицинов путем их комплексирования с белковым фактором иммунитета Im9 (молекулярная масса 9,5 кДа). Фактор Im9 соэкспрессируется с колицином и с высоким сродством связывается с колицином [126].
ПКС у бактерий наблюдается при заражении фагом. В этом плане в наибольшей мере исследована система E. coli – Т-четные фаги. Подобно эукариотическим инфицированным клеткам, гибнущим, чтобы локализовать опасный для всего многоклеточного организма патоген, некоторые штаммы E. coli несут гены, вызывающие гибель клетки после внедрения фага Т4 [123, 127, 128]. Так, ген lit блокирует синтез всех клеточных белков в ответ на экспрессию генов фага Т4. Ген lit кодирует протеазу, расщепляющую фактор элонгации EF-Tu, необходимый для синтеза белка на рибосомах [129]. Ген prrC кодирует нуклеазу, расщепляющую лизиновую тРНК. Нуклеаза активируется посредством пептидного продукта гена stp фага Т4. Гены rex вызывают у клеток, инфицированных фагом Т4, формирование трансмембранных ионных каналов, обрекающих эти клетки на гибель, если только фаг не закрывает каналы своими белками, продуктами генов rII [127, 130].
Гены, отвечающие за гибель клетки в ответ на внедрение вирулентного фага, локализуются в плазмидах или в геноме фагов (экспрессируясь в лизогенных клетках [127]). Поэтому представляется вероятным, что "альтруистические" гены, будучи подвижными и легко утрачиваемыми генетическими элементами, функционируют только у части бактериальной популяции.
ПКС представляет собой нормальную составную часть процесса развития многих прокариот. Агрегация клеток миксобактерий с формированием плодового тела со спорами сопряжена с гибелью значительной части клеточной популяции [118]. При спорообразовании у бацилл отмирает вегетативная клетка, внутри которой и созревает спора [118, 131].
Микробиологам знакома проблема появления у грам-отрицательных бактерий жизнеспособных, но некультивируемых (покоящихся) форм [132]. Такие клетки устойчивы к воздействию повреждающих агентов, малоактивны в метаболическом отношении и не размножаются. Однако они могут быть выведены из состояния покоя различными способами, специфичными для конкретных видов и штаммов бактерий, например, путем перевивки через чувствительное к патогену животное или с помощью сигнальных веществ, выделенных активно растущими клетками. Так, некультивируемые формы преобладают в популяции Micrococcus luteus после длительного голодания. Добавление 20–30% надосадочной жидкости из клеточной суспензии, выращенной на богатой среде до ранней стационарной фазы, обеспечивает активный рост некультивируемых форм [132, 133].
Вопрос о физиолого-биохимических механизмах формирования некультивируемого, но жизнеспособного состояния у бактериальных клеток остается дискуссионным, однако обращает на себя внимание сходство некультивируемых форм бактерий и эукариотических клеток на начальных стадиях апоптоза. Подобно последним, бактериальные клетки, переходя в некультивируемое состояние, уменьшаются в размерах, у них активируются протеолитические ферменты, РНК и рибосомы подвергаются деградации. Эти процессы гипотетически рассматриваются как проапоптоз, т.е. эволюционный предшественник апоптоза [118].
Активные формы кислорода (АФК) способствуют образованию некультивируемых форм в бактериальных популяциях [118]. Элиминацией АФК ведают ферменты супероксиддисмутаза, каталаза и пероксидазы. У мутантов E. coli, лишенных этих ферментов, в условиях нехватки питательных веществ наблюдали повышенные концентрации окисленных (карбонилированных) функционально важных белков, отвечающих за элонгацию полипептидной цепи при рибосомальном синтезе белка (фактор EF-G), укладку полипептидов (DnaK), поддержание архитектуры ДНК (щелочная форма белка H-NS). Лишенные супероксиддисмутазы мутанты вообще не могут быть культивированы после периода голодания, а лишенные каталазы могут быть возвращены к жизни только при культивировании в анаэробных условиях [134].
Известно, что АФК индуцируют апоптоз у животных, а антиоксиданты ингибируют этот процесс. Дрожжи погибают при воздействии пероксидом водорода (в низких концентрациях) или истощении внутриклеточного фонда глутатиона [135]. При ПКС, вызванной инсерцией гена bax, наблюдаетсянакопление АФК в клетках дрожжей; снижение их концентрации, например, путем инкубации клеток в условиях гипоксии, предотвращает bax-индуцированную ПКС у Saccharomyces cerevisiae [135]. Bax-Индуцированная гибель дрожжей выражена в большей степени в условиях дыхания, чем в условиях брожения [116]. Эти данные подкрепляют гипотезу о некультивируемом состоянии бактериальных клеток как эволюционном предшественнике апоптоза, который первоначально (на ранних стадиях эволюции) обходился без каспаз, но реализовался при участии активных форм кислорода.
Как уже отмечалось, процессы дифференцировки эукариот могут наносить существенный ущерб клеткам – в качестве иллюстрации безъядерные эритроциты млекопитающих. Сходные эффекты – при дифференцировке прокариот, осуществляемой в интересах всей клеточной популяции [118]. Так, гетероцисты цианобактерий лишены фотосистемы II, они выполняют узкоспециализированную функцию – фиксируют N2 из атмосферы – и погибают, как только отпадает потребность в азотфиксации. Узкую специализацию имеют и бактероиды, образуемые корневыми симбионтами растений – бактериями рода Rhizobium, также фиксирующими азот атмосферы.
В заключение напрашивается параллель между апоптозом и его аналогом, альтруистической гибелью клеток, и сходными биосоциальными явлениями, наблюдаемыми в популяциях многоклеточных организмов, включая человека. Не представляет ли собой программируемая клеточная смерть эволюционную предтечу того, что воспето во многих поэмах и отражено в словах "Варшавянки": "В битве великой не будут забыты павшие с честью во имя идей"?
Заключение
Программируемая клеточная гибель – механизм, широко распространенный в различных царствах живого, включая прокариот. Эволюционно ПКС возникла у прокариот как механизм противовирусной защиты популяций и была закреплена у одноклеточных эукариот. В дальнейшем, с появлением многоклеточных организмов, механизм совершенствовался и был приспособлен, наряду с защитой от патогенов, для реализации важных жизненных функций – дифференцировки клеток и тканей при эмбриогенезе и постэмбриональном развитии, элиминирования клеток иммунной системы, невостребованных, состарившихся клеток либо клеток, подвергшихся воздействию мутагенных факторов. Апоптоз у животных и человека стал неотъемлемым инструментом для осуществления наследственного и приобретенного, гуморального и клеточного ответа иммунной системы. Исследования в области апоптоза открывают новые перспективы в клеточной биологии и иммунологии.
Работа поддержана Российским фондом фундаментальных исследований (грант № 98-04-48226) и фондом "Фундаментальное естествознание" Минобразования РФ.
Список литературы
Greenberg, J. T. (1996) Proc. Natl. Acad. Sci. USA., 93, 12094-12097.
Vaux, D. L. and Korsmeyer, S. J. (1999) Cell, 96, 245-254.
Новожилова А.П., Плужников Н.Н., Новиков В.С. (1996) В кн. Программированная клеточная смерть (Под ред. В. С. Новикова.), Наука, Спб., c. 9-29.
Kerr, J.F.R., Wyllie, A.Н., and Currie, A. R. (1972) Brit. J. Cancer, 26, 239-257.
Cohen, J. J. (1993) Immunol. Today, 14, 126-130.
Thompson, C.B. (1995) Science, 267, 1456-1462.
Jacobson, M. D., Weil, M., and Raff, M. C. (1997) Cell, 88, 347-354.
Nash, P.B., Purner, M.B., Leon, R.P., Clarke, P., Duke, R.C., and Curiel, T.J. (1998) J. Immunol., 160, 1824-1830.
Галанкин В.Н., Токмаков А.М. (1991) Проблемы воспаления с позиций теории и практики. М., УДН, 120 с.
Male, D., Cooke, A., Owen, M., Trowsdale, J., and Champion, B. (1996) Advanced Immunology. Mosby, London.
Oliver, F.J., Menissier-de Murcia, J., and de Murcia, G. (1999) Am. J. Hum. Genet., 64, 1282-1288.
Tsujimoto Y. (1997) Cell Death Differ., 4, 429-434
Nicotera, P. and Leist, M. (1997) Cell Death Differ., 4, 435-442.
Leist, M., Single, B., Castoldi, A.F., Kuhnle, S., and Nicotera, P. (1997) J. Exp. Med., 185, 1481-1486.
He, C.-J., Morgan, P. W., and Drew, M. C. (1996) Plant Physiol., 112, 463-472.
Greenberg, J. T. (1997) Annu. Rev. Plant Physiol. Plant Mol. Biol., 48, 525-545.
Pennell, R. I. and Lamb, C. (1997) Plant Cell, 9, 1157-1168.
Wojtaszek, P. (1997) Biochem. J., 322, 681-692.
Fukuda, H. (1997) Plant Cell., 9, 1147-1156.
Groover, A., DeWitt, H., and Jones, A. (1997) Protoplasma, 196, 197-211.
Heath, M. C. (1998) Eur. J. Plant Physiol., 104, 117-124.
Del Rio, L. A., Pastori, G. M., Palma, J. M., Sandalio, L. M., Sevilla, F., Corpas, F. J., Jimenez, A., Lopez-Huertas, E., and Hernandez, J. A. (1998) Plant Physiol., 116, 1195-1200.
Fath, A., Bethke, P.C., and Jones, R.L. (1999) Plant J., 20, 305-315.
Levine, A., Pennell, I., Alvarez, M. E., Palmer, R., and Lamb, C. (1996) Curr. Biol., 6, 427-437.
Boyes, D. C., McDowell, J. M., and Dangl, J. L. (1996) Curr. Biol., 6, 634-637.
Jabs, T. (1999) Biochem. Pharmacol., 57, 231-245.
Bolwell, G.P., Butt, V.S., Davies, D.R., and Zimmerlin A. (1995) Free Rad. Sci., 23, 517-532.
Lane, B.G., Dunwell., J.M., Ray, J.A., Schmitt, M.R., and Cuming, A.C. (1993) J. Biol. Chem., 268, 12239-12242.
Braidot, E., Petrussa, E., Vianello, A., and Macri, F. (1999) FEBS Lett., 451, 347-350.
Mittler, R. and Lam, E. (1996) Trends Microbiol., 4, 10-15.
Deom, C.M., Lapidot, M., and Beachy, R.N. (1992) Cell, 69, 221-224.
Jacobson, M. D., Burne, J. F., and Raff, M. C. (1994) EMBO J., 13, 1899-1910.
Cohen, G. M. (1997) Biochem. J., 326, 1-16.
Thornbery, N.A. and Lazebnik, Y. (1998) Science, 281, 1312-1316.
Куцый М.П., Кузнецова Е.А., Газиев А.И. (1999) Биохимия, 64, 149-163.
Kidd, V.J. (1998) Annu. Rev. Physiol., 60, 533-573.
Solomon, M., Belenghi, B., Delledonne, M., Menachem, E., and Levine, A. (1999) Plant Cell, 11, 431-443.
Xiang, J., Chao, D. T., and Korsmeyer, S. J. (1996) Proc. Natl. Acad. Sci. USA, 93, 14559-14563.
McCarthy, N. J., Whyte, M.K.B., Gilbert, C. S., and Evan, G. I. (1997) J. Cell. Biol., 136, 215-227.
Liu, X., Zou, H., Slaughter, C., and Wang, X. (1997) Cell, 89, 175-184.
Sahara, S., Aoto, M., Eguchi, Y., Imamoto, N., Yoneda, Y., and Tsujimota, Y. (1999) Nature, 401, 168-173.
Zamzami, N., and Kroemer, G. (1999) Nature, 401, 127-128.
Green, D. S. (1998) Cell, 94, 695-698.
Raff, M. (1998) Nature, 396, 119-122.
Kumar, S. and Colussi, P.A. (1999) Trends Biochem. Sci., 24, 1-4.
Aravind, L., Dixit, V.M., and Koonin, E.V. (1999) Trends Biochem. Sci., 24, 47-53.
Nagata, S. (1997) Cell, 88, 355-365.
Golstein, P. (1997) Curr. Biol., 7, R750-R753.
Peter, M. E., Heufelder, A. E., and Hengartner, M. O. (1997) Proc. Natl. Acad. Sci. USA, 94, 12736-12737.
Green, D.R. (1998) Nature, 396, 629-630.
Ashkenazi, A. and Dixit, V.M. (1998) Science, 281, 1305-1308.
Huppertz, B., Frank, H.-G., and Kaufmann, P. (1999) Anat. Embryol., 200, 1-18.
Hengartner, M. (1998) Science, 281, 1298-1299.
Muzio, M., Stockwell, B.R., Stennicke, H.R., Salvesen, G.S., and Dixit, V.N. (1998) J. Biol. Chem., 273, 2926-2930.
Kroemer, G. (1997) Nature Med., 3, 614-620.
Adams, J.M. and Cory, S. (1998) Science, 281, 1322-1326.
Reed, J.C., Jurgensmeier, J.M., and Matsuyama, S. (1998) Biochim. Biophys. Acta, 1366, 127-137.
Gross, A., McDonnell, J.M., and Korsmeyer, S.J. (1999) Genes and Dev., 13, 1899-1911.
Mьller, W.E.G. (1999) Biol. Cell, 91, 535-565.
Kroemer, G., Zamzami, N., and Susin, S. A. (1997) Immunol. Today, 18, 44-51.
Green, D.R. and Reed, J. (1998) Science, 281, 1309-1312.
Bernardi, P., Basso, E., Colonna, R., Costantini, P., Di Lisa, F., Eriksson, O., Fontaine, E., Forte, M., Ichas, F., Massari, S., Nicolli, A., Petronilli, V., and Scorrano, L. (1998) Biochim. Biophys. Acta, 1365, 200-206.
Skulachev, V.P. (1998) FEBS Lett., 423, 275-280.
Похожие работы
... и в конечном итоге достигает ядра, где и происходит включение программы клеточного самоубийства путем активации летальных и/или репрессии антилетальных генов. Однако существование ПКС (программируемая клеточная смерть) в безъядерных системах (цитопластах – клетках, лишенных ядра) показывает, что наличие ядра не является обязательным для реализации процесса ]. Применительно к клеткам животных и ...
... Полом Нерсом (Paul M. Nurse) и американцем Лиландом Хартуэллом (Leland H. Hartwell) в 2001 году получили нобелевскую премию по физиологии и медицине за открытие генетических и молекулярных механизмов регуляции клеточного цикла – процесса, который имеет важнейшее значение для роста, развития и самого существования живых организмов Контрольные точки клеточного цикла 1. Точка выхода из G1‑ ...
... и секретировать интерлейкин-2. А этот цитокин известен своей способностью резко активизировать клетки-киллеры. Д. Иммунодефицитные состояния (ИДС) Наиболее распространенной формой патологии иммунной системы является иммунологическая недостаточность, или, согласно международной терминологии, иммунодефицитные состояния (ИДС). В основе ИДС лежат нарушения генетического кода (или других ...
... выполняет фактор стволовых клеток (SCF). На дальнейших этапах дифференцировка клеток определяется очень многими факторами, выполняющими роль как индуктора, так и факторов выживания. Существуют две альтернативные формы ответа различных популяций клеток иммунной системы на антигенную стимуляцию – пролиферация или апоптоз. Апоптоз – активная форма реакции ИКК не только на неблагоприятные, но и на ...
0 комментариев