1. Транспорт цисплатина 2
2. Внутрішньоклітинна мішень цисплатина 4
3. Вплив цисплатина на клітинний цикл та індукція апоптозу 5
4. Механізми резистентності пухлинних клітин до цисплатина 7
4.1. Механізми клітинної резистентності на
рівні цитоплазматичної мембрани 7
4.2. Внутрішньоклітинні тіолові детоксикуючі системи 9
4.2.1. Система глутатіону 9
4.2.2. Металотеонеіни 10
4.3. Репарація пошкоджень ДНК 11
4.4. Зміни генома, що асоційовані з резистентністю
до цисплатина 15
Список використаних джерел 21
Цисплатин є одним з найбільш загальновживаних препаратів у лікуванні солідних пухлин людини. Однак ефективне застосування цисплатина у клініці часто лімітується токсичністю препарату та розвитком резистенстності до нього. Резистентність до цисплатина має комплексний характер і пов`язана з рядом особливостей пухлинних клітин, включаючи зміни в проникності цитоплазматичної мембрани, підвищення активності детоксикуючих та репаративних систем клітини, порушення експресії генів fos, nm23, p53, mdm2, bcl-2 та інші. Порушення біохімічних сигнальних шляхів апоптозу також можуть бути основою для розвитку резистентності. Розуміння молекулярних основ дії цисплатина та механізмів розвитку резистентності до нього дозволять значно покращити результати клінічних випробувань препарату.
1.Транспорт цисплатинаВзаємодія цисплатина з плазматичною мембраною є першою щаблею багатостадійного процесу реалізації цитотоксичного ефекту препарату. Ступінь пошкодження мембран цитостатиком залежить від текучості їх ліпідної компоненти, яка визначається швидкістю проходження процесів перекисного окислення ліпідів [1].
На сьогодні існують дві гіпотези поглинання препарату пухлинною клітиною. Більш аргументована гіпотеза пасивного транспорту, основана на фактах відсутності інгібування переносу цисплатина за допомогою його структурних аналогів та проникнення цисплатина в клітину без насичення до межі його розчинності у культуральному середовищі [2]. Але ряд вчених відстоює ідею існування активного транспортера цисплатина. У 1984 році був відкритий інгібітор синтезу протеінів бензальдегід, який послаблював цитотоксичний ефект цисплатина за рахунок зменшення його накопичення в клітині. Інший відомий інгібітор білкового синтезу – циклогексамід не мав такого ефекту. Це й стало основою для заключення, що бензальдегід якимось чином безпосередньо реагує з мембранним білком-транспортером, уповільнюючи таким чином проникнення цисплатина у клітину [3].
Піздніше було знайдено, що альдегідні похідні пиридоксаль та пиридоксаль-5-фосфат також значно послаблюють цитотоксичну активність цисплатина. Відомо, що ці речовини утворюють основи Шиффа з аміногрупами на поверхні клітини. Було показано, що і бензальдегід і інші похідні інгібують проникнення цисплатина у клітину на 50% у порівнянні з контролем. Є дані про те, що попередня інкубація клітин з інгібітором Nа/К АТФ-ази уабаіном також зменшує проникнення цисплатина у клітину на 50%. Подальші дослідження у цьому напрямку показали, що безпосередньо Nа/К АТФ-аза не є транспортером цисплатина, однак транспорт ліків Na-залежний, а також залежить від мембранного потенціалу клітини [4,5].
Розглянемо загальновживану модель акумулювання цисплатина в клітині, яка влаштовує прибічників обох гіпотез. Швидке накопичення половини цисплатина у клітині проходить за рахунок пасивної дифузії, тоді як інша половина транспортується через канали, що закриваються. Якщо активність останніх заблоковано, то можна очикувати зменшення проникнення цисплатина в клітину на 50%. Дані про регуляцію активності каналів дозволяють зробити висновок, що проходження цисплатина через них регулюється каскадом реакцій фосфорилювання, що ініціюються протеінкіназою А (РКА), протеінкіназою С (РКС) чи кальмодулінзалежними кіназами [3]. Здатність мембранонепроникних альдегідних похідних блокувати накопичення цисплатина у клітині на 50% обумовлюється реактивністю зовнішніх аміногруп білків, що формують такий канал. Той факт, що інгібування початкового потоку цисплатина у клітину на 50% відбувається за допомогою уабаіна, підтверджує, що повний мембранний градієнт, зумовлений Na/K АТФ-азою, важливий для роботи каналів.
Bиведення цисплатина з клітини – двохфазний процес з дуже короткою почaтковою фазою тривалістю 5 хв та більш довгою кінцевою фазою [6,7]. Встановлено існування АТФ-залежного переносника цисплатина кон`югованого з глутатіоном [8,9]. Описана АТФ-залежна глутатіон-S-кон`югат експортуюча помпа (ГS-Х-помпа) , що має широкий спектр субстратної специфічності та транспортує органічні аніони, лейкотрієн С4 та інші сполуки, що несуть великі гідрофобні ділянки та хоча б два від`ємні заряди. ГS-Х-помпа виводить потенційно токсичні кон`югати глутатіон-S-платина (ГS-Pt) з пухлинних клітин, таким чином беручи участь в формуванні резистентності клітин до цисплатина [10].
2.Внутрішньоклітинна мішень цисплатина.
Внутрішньоклітинною мішенню цисплатина є ДНК, з якою препарат ковалентно зв`язується. Біфункціональні продукти взаємодії цисплатина з ДНК, що називаються цисплатин-ДНК-адукти, блокують реплікацію, транскрипцію і, як наслідок, - клітинну проліферацію. Цисплатин діє на 7-му позицію залишка гуаніна та формує декілька типів адуктів з основами ДНК. Два основні адукти це G-G внутрішньоланцюгові зшивки (складають 60-65% усіх адуктів), A-G внутрішньоланцюгові зшивки ( 20-25% усіх адуктів), а також міжланцюгові та ДНК-білкові зшивки [11]. Після дії цисплатина основна кількість адуктів утворюються вже через 6-12 годин [12]. Формування адуктів відбувається в два етапи. Спочатку має місце швидкий етап алкілування: один ланцюг ДНК формує цисплатин-моноадукт. Далі повільна фаза: реакція з`єднання з другим ланцюгом.
Більшість робіт у напрямку дослідження взаємодії цисплатина та ДНК стосується ядерної ДНК. Але в останні роки вчених зацікавила у цьому відношенні мітохондріальна ДНК. Виявилось, що ця ДНК у 4-50 разів (для різних модельних систем) більш чутлива до пошкоджуючого ефекту цисплатина, ніж ядерна ДНК [13,14]. А у зв`язку з сучасними уявленнями про роль мітохондрій у реалізації програми апоптозу цей факт набуває нового значення.
3.Вплив цисплатина на клітинний цикл та індукція апоптозу.
Відомо, що у багатьох модельних системах in vitro цисплатин не э фазоспецифічним протипухлинним препаратом. Пухлинні клітини у різних фазах клітинного циклу однаково чутливі до нього [15]. Але дані деяких досліджень свідчать про те, що все таки у фазі G2/M клітини є більш чутливими до дії цитостатика [16]. Низькі концентрації цисплатина у клітинах викликають уповільнене проходження S-фази і зупинку у G2/M-фазі клітинного циклу [17]. В залежності від концентрації препарату і, відповідно, пошкодження ДНК, клітина після G2/M-зупинки вступає в наступну фазу клітинного циклу – мітоз, або входить в апоптоз. Щодо дії цитостатика на регулятори клітинного циклу, то встановлено, що цисплатин не впливає на внутрішньоклітинний вміст цикліну А [18]. Але при дії препарату збільшуються рівні цикліну В, p34cdc2 а також зростає активність гістон Н1-кінази [19]. Після дії цисплатина підвищується експресія цикліну D1, cdk4, а також збільшується рівень фосфорилювання білка ретинобластоми, тобто з`являються усі ознаки руху клітини по клітинному циклу [20]. І дійсно, якщо обробляти цисплатином клітини, що знаходяться у стані спокою (G0), вони виходять у G1-фазу, повільно проходять S-фазу і зупиняються у G2/M-фазі клітинного циклу.
Після пошкодження ДНК цисплатином у клітинах відбувається збільшення експресії білка Р53 і Р53-залежне підвищення експресії білка р 21, який і спричиняє зупинку клітинного циклу [21].
Субтокичні дози цисплатина індукують загибель клітин за типом апоптозу, з усіма характерними біохімічними та морфологічними ознаками процесу: екстерналізацією фосфотидилсерину, активацією каспаз, фрагментацією ДНК, утворенням апоптичних тілець [22-24].
В багатьох модельних системах in vitro показано, що лікарські протипухлинні препарати, в тому числі і цисплатин, викликають підвищення експресії поверхневого рецептора Fas, що потребує синтезу білка de novo [25], а також його ліганда FasL [26]. Але на сьогодні вважається, що індукований проитипухлинними ліками апоптоз не є залежним від активації безпосередньо Fas-системи, оскільки вживання антагоністичних анти-CD95 моноклональних антитіл не впливало на розвиток апоптозу під дією цитостатиків [27].
Особливо цікавими є дані щодо ефекторних механізмів апоптозу, індукованого цисплатином. Відомо, що у цьому процесі бере участь каспаза-3, оскільки застосування її інгібіторів білка CrmA та Z-VAD-CH2DCB призводить до гальмування апоптозу, спричиненого цитостатиком [28,29]. Каспаза-3 активує інші каспази, крім того розщіпляє фактор ініціації трансляції 4G (eIF4G), що призводить до гальмування синтезу білка [30].
4.Механізми резистентності пухлинних клітин до цисплатина.
4.1.Механізми клітинної резистентності на рівні цитоплазматичної мембрани
Одним з основних механізмів клітинної резистентності до цисплатина розглядають особливості будови цитоплазматичної мембрани та прямого та зворотнього транспорту крізь неї.
Однією з особливостей пухлинних клітин, резистентних до дії цисплатина, є зменшена у порівнянні з чутливими клітинами акумуляція препарату [31,32].
Дана модель пояснює, чому так багато клітинних ліній резистентних до дії цисплатина відрізняються зменшеним накопиченням цисплатина. Мутації, що призводять до змін структури каналів чи гіперполяризації клітинної мембрани, можуть спричинити виникнення резистентного клона клітин.
Нещодавно до клітин мишиної лімфоми R1.1, резистентних до дії цисплатина були одержані антитіла, які реагували з глікопротеідом з молекулярною масою 200кД, гіперекспресованим на поверхні цих клітин. Кількість білка на клітинній поверхні знаходилась у зворотній залежності від акумулювання цисплатина у клітинах та корелювала з рівнем їх чутливості до цисплатина. Було запропоновано, що даний білок може бути аналогом Р-глікопротеіда (Pgp) і є непрацездатним каналом, що гіперекспресований на клітинній поверхні для компенсації дефектної функції [33].
В резистентних до цисплатина клітинах з підвищеним вмістом глутатіона часто спостерігається гіперекспресія ГS-Х-помпи. В цитоплазматичних везикулах таких клітин знаходиться більше кон`югатів ГS-Pt, ніж в чутливих клітинах [34]. Така внутрішньоклітинна компартменталізація ліків та продуктів їх метаболизму також вважається одним з механізмів лікарської резистентності. В багатьох випадках резистентні пухлинні клітини відрізняються розвинутою везикулярною сіткою, що дозволяє їм просторово нейтралізувати токсини та виводити їх шляхом екзоцитозу.
В експериментах з використанням бутионінсульфоксиміна (BSO), інгібітора синтезу глутатіона, було показано, що при витощенні внутрішньоклітинного глутатіона активність ГS-Х-помпи лише трохи зменшується [35]. Таким чином, ефективність транспорту ГS-Pt-кон`югатів визначається не тільки концентрацією останніх, а й експресією переносника.
Останнім часом з`явилися роботи, що засвідчують, що білок, асоційований з загальною лікарською резистентністю (MRP але не Pgp) може функціонувати як ГS-Х-помпа [36,37]. Ця гіпотеза підтверджується такими фактами: 1) клітини, що гіперекспресують MRP секретують у культуральне середовище більше глутатіона; 2) витощення внутрішньоклітинного глутатіона за допомогою BSO запобігає виведенню ліків з клітин за допомогою MRP; 3) гіперекспресія MRP відповідає підсиленому АТФ-залежному транспорту кон`югатів ГS-Х; 4) антитіла до MRP реагують з білком з молекулярною масою 190 кД, який може зв`язуватись з ГS-кон`югатами та лейкотрієном С4.
Таким чином, видалення з клітин комплексів глутатіон-S-платина переносниками з активністю ГS-Х-помпи є важливим механізмом резистентності, завдяки якому внутрішньоклітинна концентрація препарату підтримується на низькому рівні, що сприяє зменшенню його пошкоджуючого ефекту.
4.2.Внутрішньоклітинні тіолові детоксикуючі системи 4.2.1. Система глутатіонаГлутатіон та ферменти його метаболізму є важливими елементами меxанізму резистентності клітин до алкілуючих агентів, в тому числі сполук платини.
Більшість клітинних ліній, резистентних до дії цисплатина відрізняються підвищеним вмістом внутрішньоклітинного глутатіона та/чи гіпреактивністю фермента, що ініціює синтез глутатіона g-глутамілцистеінсинтетази (g-ГЦС) [38-40]. Однак описані приклади стійких до дії цисплатина клітинних ліній з нормальним [41,42] та зниженим [43] у порівнянні з чутливими лініями вмістом глутатіона.
Сучасні стереоскопічні та спректроскопічні методи дозволили визначити, що глутатіон та цисплатин реагують безпосередньо у безклітинній системі в молярному співвідношенні 2/1, утворюючи хелатний комплекс диглутатіонплатини. Після 12 годин інкубації клітин в середовищі з цисплатином концентрація ГS-Pt-кон`югатів стає максимальною. При цьому 60% внутрішньоклітинної платини знаходиться у зв`язаному стані [10].
Заслуговують на увагу дані, отримані в експериментах з використанням інгібітора синтезу глутатіона бутіонінсульфоксиміна (BSO). Обробка BSO клітинних ліній раку шлунка людини MKN-28 та MKN-45 , раку яєчника КК та МН, аденокарциноми ротової порожнини КВ призводить до сенсибілізації клітин до дії цисплатина [44,45]. Причому витощення внутрішньоклітинного глутатіона за допомогою BSO не впливало на акумулювання цисплатина в клітинах а також формування кон`югатів GSH-Pt [46].
При вивчeнні взаємодії ферментів метаболізму глутатіона та їх ролі в механізмах резистентності велике значення мають експерименти з використанням культур клітин, трансфекованих генами відповідних ензимів. Наприклад, клітини раку легенів людини, трансфековані геном фермента g-ГЦС, мають у 2 рази більше глутатіона, в 1,6 рази підвищену активність ГS-Х-помпи та в 1,5 рази меньшу акумуляцію цисплатина, в 6,7 рази більшу резистентність до цисплатина у порівнянні з батьківською клітинною лінією. Витощення внутрішньоклітинного глутатіона в трансфектах за допомогою BSO не впливало на резистентність до цисплатина. У таких випадках зберіглась висока активність ГS-X-помпи і тому внутрішньоклітинна концентрація платини не змінилася. Таким чином, в даній клітинній системі резистентність обумовлена посиленим видаленням ГS-Pt-конюгатів за допомогою ГS-Х-помпи, що не загубила своєї активності навіть при витощенні субстрата (глутатіона) [47].
В той же час не винайдено ніякого зв`язку між рівнем експресії глутатіонредуктази та глутатіонпероксидази та ступенем стійкості клітин до дії цисплатина [48,49].
4.2.2.Металотеонеіни
Особлива роль у формуванні резистентності відводиться металотеонеінам.
Оскільки у вільному стані ці сполуки нуклеофільні, металотеонеіни зв`язуються з електрофільними протипухлинними препаратами типу цисплатина, а також мелфаланом та деякими антибіотиками: адріаміцином, блеоміцином та доксорубіцином.
Клітини, що гіперексперсують металотеонеіни, часто резистентні до дії цисплатина [50,51]. Однак металотеонеін не є обов`язковим компонентом резистентності до цього препарату, оскільки зустрічаються резистентні до цисплатина клітинні лінії, в яких металотеонеін не виявляється [43].
При обробці цисплатином резистентних до препарата клітин з підвищеним вмістом металотеонеіна 70% внутрішньоклітинної платини знаходять у зв`язаному з білком стані.
Досліди по трансфекції гена металотеонеіна ІІа показали, що клітини-трансфекти набувають резистентності до цисплатина, хлорамбуцила та мелфалана [52].
Добре виражена експресія металотеонеіна в пухлинах може мати прогностичне значення. Наприклад, при лікування хворих на рак стравоходу за допомогою цисплатина 5-річна життєздатність пацієнтів з металотеонеін-від`ємними пухлинами становить 56%, а пацієнтів з металотеонеін-позитивними пухлинами – 26% [53].
З наведених вище даних можна заключити, що у випадках підвищеної експресії металотеонеін є важливою складовою резистентності до цисплатина.
4.3.Репарація пошкоджень ДНК.
Резистентність клітин до дії цисплатина може залежати від стану системи репарації пошкоджень ДНК.
Один з механізмів резистентності клітин до цисплатину – прискорена репарація цисплатин-ДНК-адуктів [54]. Чутливі до дії цисплатина клітинні лінії відрізняються зниженою здатністю ліквідувати основні адукти ДНК та цисплатина (GG-Pt та GA-Pt), а також послабленою активністю ДНК-полімераз [55,56]. Обробка резистентних до цисплатина клітин афідикоіном – інгібітором ДНК-полімераз a та b повертає чутливість до цисплатина, що стверджує роль репарації адуктів цисплатин-ДНК у формуванні резистентності [57].
На сьогодні мало відомо, щодо ліквідації продуктів платинування ДНК у мітохондріях. Вважають, що мітохондріальна ендонуклеаза G (Endo G) може брати участь у репаративних процесах цих органел [58].
Нещодавно з культуральної рідини пухлинних клітин та біопсійного матеріалу пухлин людини були виділені білки HMG (high-mobility group) та їм подібні, які зв`язуються з ДНК, що пошкоджена цисплатином, але не трансплатином чи ультрафіолетовим випроміненням [59,60]. Ці білки – висококонсервативні, локалізуються у цитоплазмі та ядрі клітини. Їх біологічна роль ще точно не встановлена. Інтенсивність зв`язування ДНК з цисплатином та HMG-подібними білками прямо пропорційна ступеню пошкодження ДНК. ДНК резистентних клітин зв`язується з цими білками більш ефективно. HMG-білки взаємодіють з платинованою ДНК, закривають пошкоджені сайти від ферментів ексцизійної репарації. Припускають, що зв`язування HMG-подібних білків з пошкодженою ДНК може викликати зупинку реплікації чи транскрипції, а також впливати на роботу систем контролю клітинного циклу при руйнуванні ДНК.
Відомо, що такі дефекти системи репарації невідповідностей (mismatch repair), як недостатня експресія білків hMSH2 чи hMLH-1, призводять до виникнення резистентності до цисплатина [61,62]. MSH2-білок сам по собі чи разом з білком GTBP/p160 розпізнає невеликі зміни у ланцюгу ДНК, наприклад, продукти платинування ДНК, і зв`язується з ними.
Априорі можна було б припустити, що відсутність якої-небудь частини системи репарації ДНК сприяє розвитку гіперчутливості до дії цисплатина, що відбувається у клітинах, дефектних за системою ексцизійної репарації. Але у випадку порушення функції системи mismatch repair клітина набуває резистентності до цисплатина. Цей феномен можна розглядати з двох позицій. По-перше, порушення системи mismatch repair може бути наслідком індукованого цисплатином випадкового мутагенезу, що в результаті обумовлює стійкість до дії цисплатина. По-друге, саме дефект у роботі цієї системи репарації може покласти початок резистентності [63,64].
Комплекс білків репарації “розпізнає” адукти у ланцюгу-шаблоні ДНК та намагається виправити ланцюг, що синтезується [65]. Так, поки існує адукт у ланцюгу-шаблоні, а підбір нових основ не ліквідує невідповідність, що існує, генерується сигнал, який запускає програму апоптозу. Якщо система mismatch repair не працює, такий сигнал не генерується, клітини стають толерантними до адуктів цисплатин-ДНК та набувають резистентного фенотипу.
У випадку порушення системи ексцизійної репарації спостерігається протилежний ефект. Клітини, дефектні по системі ексцизійної репарації значно більш чутливі до дії цисплатина, ніж нормальні клітини [66].
Показано, що цисплатин індукує експресію важливого для ексцизійної репарації гена ERCC-1. Але до активації гена ERCC-1 відбувається активації генів c-fos та c-jun, а також фосфорилювання білка c-Jun [67].
Нещодавно був відкритий новий ген BRCA1, повязаний з репарацією пошкодженої ДНК. Показано, що гіперекспресія цього гена в клітинах рака яєчника та молочної залози асоційована з резистентністю до дії цисплатина [68].
Топоізомерази І та ІІ, що релаксують спіралі ДНК, є важливими ферментами реплікації, транскрипції та рекомбінації. Вони відіграють суттєву роль у процесах усунення адуктів ДНК-цисплатин. У багатьох резистентних до дії цисплатина клітинах спостерігається підвищена активність топоізомерази ІІ [69]. Інгібітори топоізомераз І та ІІ: етопозид, камтотецин, СРТ-11, SN-38, новобіоцин – викликають сенсибілізацію резистентних клітин до дії цисплатина [70-72].
Ще одним механізмом резистентності до цисплатина на рівні ДНК може бути підвищена концентрація в клітині вільних нуклеотидів (наприклад, АТФ, АДФ), які конкурують з ДНК за зв`язування з цисплатином [73]. Таким чином, вільні нуклеотиди, концентрація яких у цитоплазмі відрізняється в різних клітинах, можуть значно впливати на чутливість пухлин до цисплатина.
Важливо також розглянути утворення зшивок ДНК-білок під дією цисплатина. Хоча кількість цих адуктів набагато менше, ніж у разі між- та внутри- ланцюгових зшивок, але відмічено, що вони також відповідальні за цитотоксичний ефект цисплатина. Наприклад, було показано, що одна резистентна до дії цисплатина клітинна лінія раку яєчника утримувала у 6 разів менше цитокератина 18, ніж чутлива лінія. Трансфекція кДНК цитокератина 18 в клітини резистентної лінії давала клони з підвищеним рівнем цього білка та у більшості випадків з чутливим фенотипом. При цьому було встановлено, що після обробки цисплатином, з ДНК у клітинах зв`язуються негістонові білки. Пізніше ці білки було ідентифіковано як цитокератини [74]. Можливо, що формування зшивок цитокератин-ДНК, асоціюється з цитотоксичністю цисплатина. Тоді зменшення продукції цитокератина 18 у резистентних клітинах веде до зменшення кількості зшивок білок-ДНК та, як наслідок, зниження чутливості до цисплатина.
Виходячи з вищенаведеного, можна заключити, що резистентність до дії цисплатина на рівні ДНК обумовлена підвищеною активністю систем репарації клітини чи її толерантністю до існуючих ДНК-Pt-адуктів.
4.4.Зміни генома, що асоційовані з резистентністю до цисплатина.
Оскільки клітинна резистентність є стійко спадковою ознакою в ряду поколінь, можна зробити висновок, що стійкість до дії цисплатина визначається генетичними особливостями клітини.
Встановлено, що основна роль в цьому феномені належить 11-й та 16-й хромосомам у людини [75]. На клітинних гібридах з різним хромосомним складом було показано, що обовязковою умовою резистентного фенотипа є наявність 16-ї хромосоми з резистентних клітин та 11-ї хромосоми з чутливих клітин. При цьому основну роль грає 16-та хромосома, а 11-та хромосома необхідна для її нормального функціонування. Ці хромосоми залучені у різні боки резистентності. На 16-тій хромосомі присутні гени MRP (multidrug resistance associated protein), LRP (lung resistance protein), гени додаткового білка ексцизійної репарації-4, металотеонеіна. На 11-їй хромосомі розташовані гени, що кодують глутатіон трансферазу p (ГSТ-p), а також протеін, що розпізнає специфічні послідовності пошкодженої ДНК.
Чутливість пухлинних клітин до дії цисплатина може залежати від функціональної активності генів р 53 та генів родини bcl.
На сьогодні все ще залишається відкритим питання про зв`язок лікарської резистентності з р 53-статусом клітин. Невизначеність тут існує тому, що роль білка Р 53 у хемочутливості клітин до лікарської терапії розглядають з двох протилежних точок зору: 1) експресія білка Р 53 дикого типу (wt p 53) збільшує чутливість до хіміотерапії шляхом прискорення входження клітин в апоптоз; 2) білок wt P 53 зменшує хемочутливість, оскільки після пошкодження ДНК обумовлює зупинку росту для репарації ДНК [76].
Відомо, що після дії цисплатина у чутливих клітинах збільшується рівень експресії гена р 53 та відповідно – білка Р 53. Р 53 індукує експресію білка р 21/WAF1/CIP1-інгібітора циклін залежних кіназ, що грає важливу роль у зупинці клітинного циклу у G1-фазі після генотоксичного стресу [77,78]. Під час цієї зупинки клітиною приймається “рішення”: репарувати ДНК чи входити в апоптоз, в залежності від глибини пошкодження. На сьогодні ще невідомі усі біохімічні подробиці того, як активація р 53 ініціює апоптоз.
Більшість робіт по вивченню функціональної активності Р 53 у резистентних до дії цисплатина клітинах засвідчують, що мутації гена р 53 призводять до розвитку резистентності до хіміотерапії [79-82]. Показано, що в резистентних клітинах часто порушена внутрішньоклітинна локалізація білка Р 53 [83] а також не відбувається індукція експресії Р 53 та р 21 цисплатином [84,85]. Досліди по трансфекції гена р 53 також довели, що перенос у дефектні за функцією Р 53 чи null-клітини wt p53-гена обумовлює збільшення чутливості до лікарських препаратів, а трансфекція мутантного гена mut p 53 спричиняє розвиток резистентності [86].
Досліджено, що в клітині після дії цисплатина разом з р 53 індукується експресія гена mdm 2. Продукт даного гена відрізняється властивістю утворювати комплекси з білком Р 53 , таким чином дезактивуючи частину внутрішньоклітинного Р 53. В резистентних до цисплатина клітинах іноді спостерігається гіперекспресія гена mdm 2 [87] а використання антисмислових (проти mdm 2) нуклеотидів у такій системі призводить до збільшення чутливості до лікарської терапії [88].
Таким чином, функція Р 53 може бути інактивована двома шляхами: мутаціями безпосередньо гена р 53 а також підвищеним комплексоутворенням Р 53 з MDM 2.
Але дані деяких досліджень заперечують, що мутації р 53 обов`язково спричиняють розвиток лікарської резистентності. Feudis з співавторами показали, наприклад, на 9 клітинних лініях раку яєчника з різним р 53- статусом, що в цих клітинних лініях після обробки цисплатином рівень експерсії р 21 підвищується однаковo і немає різниці у чутливості до апоптозу, індукованого дією препарата [89,90].
Однак відомо, що більш половини злоякісних новоутворень людини відрізняються мутаціями р 53, i ця ознака є важливим показником чутливості пухлин до хіміотерапії [91]. У клінічних дослідженнях показано, що рівень експресії р 53 у зразках біопсійного матеріала при раку шлунка та яєчників може бути важливим прогностичним показником захворювання [92]. Досліджено, що Р 53-позитивний фенотип пухлин асоціюється з поганим прогнозом [93,94]. Це пояснюється тим, що мутантна форма білка має подовшений час напівжиття і тому легше виявляється імуногістохімічно.
Білки родини Bcl (Bcl-2, Bcl-Xl, Bcl-Xs, Bax та інші) грають важливу роль у регуляції процесу апоптоза. Деякі з них (Bcl-Xl, Bcl-2) гальмують розвиток апоптоза, тоді як інші (Bcl-Xs, Bax, Bak) – навпаки є промоторами цього процесу. Відомо, що білки даної родини здатні гетеродимеризуватись, утворюючи умовний “реостат”, який регулює функціональну активність білків [95]. Досліджено, що білки Bcl-2 та Bcl-Xl гіперекспресовані при багатьох неопластичних захворюваннях людини. Причому така гіпрекспресія асоційована з резистентністю пухлинних клітин in vitro до протипухлинних препаратів [96]. Така стійкість клітин пов`язана з порушенням механізмів індукції апоптозу у відповідь на лікарську терапію. У дослідах з використанням клітин траксфекованих генами bcl-2 чи bcl-xl було показано, що трансфекти набувають резистентний фенотип до цілого ряду лікарськиї препаратів включаючи цисплатин [97,98]. У таких модельних клітинах була значно зменшена деградація ДНК після обробки цисплатином. Якщо порівнювати внесок продуктів генів bcl-2 та bcl-xl у антиапоптозному ефекті, то показано, що білок Bcl-Xl має більший вплив [99].
Після обробки цисплатином у чутливих клітинах, що входять в апоптоз, не змінюється рівень експресії білків Bcl-2, Bcl-Xl та Bax (24kDa), але підвищується рівень білків Bak та Bax (21 kDa), тобто білків-агоністів апоптозау [100-102]. Відомо, що в деяких резистентних до цисплатина клітинних лініях спостерігається знижений рівень експресії Bax [103], а трансфекція гена bax спричиняє підвищення чутливості до хіміотерапії [104]. А от клітини з підвищеною експресією Bak відрізняються більшою чутливістю до дії цисплатина [105].
Відомо, що білок Bcl-2 часто гіперекспресований у пухлинах людини. Причиною такої гіперекспресії вважають часті ділеції великих нетрансльованих послідовностей з 3 та 5 – кінців гену bcl-2 (78-А). Дещо суперечливі дані імуногістохімічних досліджень експресії білків родини Bcl у зразках біопсійного матеріалу та прогнозування чутливості до лікарської терапії у порівнянні з дослідженнями in vivo. Наприклад, одні дослідники повідомляють, що Вcl-2-позитивні неоплазії мають слабку відповідь на хіміотерапію та поганий прогноз [106,107], інші – навпаки, спостерігають, що експресія Вcl-2 у пухлинах асоціюється з покращеним виживанням [108]. Експресія Вax окремо не має ніякого прогностичного значення [109].
Роботи, зроблені з використанням резистентних до дії цисплатина лініях клітин показали важливість онкогена fos в резистентності до цього препарату. В резистентних лініях клітин спостерігали підвищений рівень експресії гена fos. Були визначені дві важливі функції Fos-білка: транскрипційна активація синтеза ДНК та участь у клітнинній проліферації. Fos-білок контролює клітинну відповідь на пошкодження ДНК цисплатином через активацію генів топоізомерази І, полімерази b та металотеонеіна [2].
Повідомляється про те, що гіперекспресія супресорного гена nm 23 супроводжується, як правило, збільшенням чутливості до дії цисплатина. Дані результати отримано з досліджень in vitro на клітинних лініях карциноми молочної залози людини MDA-MB-435, лінії карциноми яєчника OKAR-3 та лінії меланоми К-1735-ТК, а також при дослідженні клінічного матеріалу пухлин молочної залози. У всіх досліджуваних системах гіперекспресія nm 23 призвела до формування у клітинах великої кількості міжланцюгових зшивок ДНК та збільшення чутливості до цисплатина [110].
Цікавими є дані по переносу резисткнтного до цисплатина фенотипу при трансфекції генів v-src. Непухлинні епітеліальні клітини людини HAG-1 після трансфекції гену v-src набували неопластичного фенотипу та резистентності до цисплатина. У трансформованих клітинах спостерігали значне зменьшення формування міжланцюгових зшивок ДНК з їх послідовним швидким видаленням. Після обробки даних клітин інгібіторами src-кіназ знижувався рівень резистентності до цисплатина. Результати цієї роботи вказують на можливість участі продукту гена v-src в індукції резитентності до цисплатина шляхом модуляції деяких шляхів репарації ДНК [111].
Ампліфікація гена сорцина (кальцій-звязуючого білка з молекулярною масою 19-22кД) в деяких модельних системах асоціювалася з резистентним фенотипом [112]. Важко зараз сказати, чи є резистентність пов`язаною саме з сорцином чи разом з геном сорцина у клітинах ампліфіковані і інші, більш важливі для розвитку резистентності гени.
Таким чином резистентність до цисплатина має комплексний характер і пов`язана з рядом особливостей клітин на рівні цитоплазматичної мемрани, внутрішньоклітинних систем детоксикації, систем репарації та порушення функціональної активності генів p 53, bcl-2, fos, mdm 2, nm23та інших.
Список літератури1. Чехун ВФ. Роль плазматичних мембран нормальних і пухлинних клітин в механізмі реалізації цитотоксичних ефектів координаційних сполук платини [Автореф дис ... докт мед наук]. Киев: ИЭПОР, 1994. 40 сс.
2. Scanlon KJ, Kashani-Sabet M, Tone T, Funato T. Cisplatin resistance in human cancers. Pharmacol Ther 1991; 52:385-406.
3. Gately DP, S.B.Howell SB. Cellular accumulation of the anticancer agent cisplatin. Br J Cancer 1993; 67:1171-6.
4. Ohmori T, Morikage T. The mechanism of the difference in cellular uptake of platinum derivatives in non-small cell lung cancer cell line (PC-14) and its cisplatin-resistant subline (PC-14/CDDP). Jpn J Cancer Res 1993; 84: 83-92.
5. Scanlon KJ, Safistein RL, Thies H, Gross RB, Waxman S, Guttenplan JB. Inhibition of amino acid transport by cis-diamminedichloroplatinum (II) derivatives in L1210 murine leukemia cells. Cancer Res 1983; 43: 4211-5.
6. Andrews PA, Velury S, Mann SC, Howell SB. Cis-diamminedichloroplatinum II accumulation in sensitive and resistant human ovarian carcinoma cells. Cancer Res 1988; 48: 68-73.
7. Richon VM, Schulte N, Eastman A. Multiple mechanisms of resistance to cis-diamminedichloroplatinum (II) in murine leukemia L1210 cells. Cancer Res 1987 47: 2056-61.
8. Zaman GJR, Lakelma J, Tellingen O, Beijnen J, Dekker H, Paulusma C, Oude Elferink RPJ, Baas F, Borst P. Role of glutathione in the export of compaunds from cells by the multidrug-resistance-associated protein. Proc Natl Acad Sci USA 1995; 92:7690-4.
9. Muller V, Meijer C, Zaman GJR. Overexpression of the gene encoding the multidrug resistance-assiciated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc Natl Acad Sci USA 1994; 91: 13033-7.
10. Ishikawa T, Ali-Osman F. Glutathione-associated cis-diamminedichloroplatinum (II) metabolism and ATP-dependent efflux from leukemia cells. J Biol Chem 1993; 268: 20116-25.
11. Laurent G, Erickson LC, Sharkey NA, Kohn KW. DNA cross-linking and cytotoxicity induced by cis-diamminedichloroplatinum (II) in human normal and tumor cell lines. Cancer Res 1981; 41: 3347-51.
12. Sorenson CM, Barry MA, Eastman A. Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J Natl Cancer Inst 1990; 82: 749-55.
13. Olivero OA, Chang PK, Lopez-larraza DM, Semino-Mora MC, Poirier MC. Preferential formation and decreased removal of cisplatin-DNA adducts in chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat Res 1997, 391(1-2): 79-86.
14. Giurgiovich AJ, Diwan BA, Olivero OA, Anderson LM, Rice JM, Poirier MC. Elevated mitochondrial cisplatin-DNA adduct levels in rat tissues after transplacental cisplatin exposure. Carcinogenesis 1997, 18(1): 93-6.
15. Gorczyca W, Gong I, Ardelt B, Traganos F, Darzynkiewicz Z. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor agents. Cancer Res 1993, 53(13): 3186-92.
16. el Alaoui S, Lawry J, Griffin M. The cell cycle and induction of apoptosis in a hamster fibrosarcoma cell line treated with anticancer drugs: its importance to solid tumor chemotherapy. J Neurooncol 1997, 31(1-2): 195-207.
17. Vaisman A, Varchenko M, Said I, Chaney SG. Cell cycle changes associated with formation of Pt-DNA adducts in human ovarian carcinoma cells with different cisplatin sensitivity. Cytometry 1997, 27(1): 54-64.
18. Nishio K, Saigo N. Effect of cisplatin on cell cycle regulators. Gan To Kagaku Ryoho 1994, 21(3): 289-94.
19. Dimanche-Boitrel MT, Micheau O, Hamman A, Haugg M, Eymin B, Chauffert B, Solary E. Contribution of the cyclin-dependent kinase inhibitor p27KIP1 to the confluence-dependent resistance of HT 29 human colon carcinoma cells. Int J Cancer 1998, 77(5): 796-802.
20. Gill JS, Mindebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest 1998, 101(12): 2842-50.
21. Megyesi J, Safirstein RL, Price PM. Induction of p 21 WAF1?CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest 1998, 101(4): 777-82.
22. Otto AM, Paddenberg R, Schubert S, Mannherz HG. Cell cycle arrest, micronucleus formation, and cell death in growth inhibition of MCF-7 breast cancer cells by tamoxifen and cisplatin. J Cancer Res Clin Oncol 1996, 122(10): 603-612.
23. Boersma AW, Nooter K, Oostrum RG, Stoter G. Quantification of apoptotic cells with fluorescein isothiocyanate-labeled annexin V in chinese hamster ovary cells cultures treated with cisplatin. Cytometry 1996, 24(2): 123-130.
24. Ormerod MG, O`Neill C, Robertson D, Helland LR, Harrap KR. Cis-diamminedichloroplatinum (II)-induced cell death through apoptosis in sensitive and resistant human ovarian carcinoma cell lines. Cancer Chemother Pharmacol 1996, 37(5): 463-471.
25. Uslu R, Jewett A, Bonavida B. Sensitization of human ovarian tumor cells by subtoxic CDDP to anti-fas antibody mediated cytotoxicity and apoptosis. Gynecol Oncol 1996, 62(2): 282-91.
26. Fulda S, Sieverts H, Friesen C, Herr I, Debatin KM. The CD95(APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res 1997, 57(17): 3823-9.
27. McGahon AJ, Costa Pereira AP, Daly L, Cotter TG. Chemotherapeutic drug-induced apoptosis in human leukaemic cells is independent of the Fas(APO-1/CD95) receptor/ligand system. Br J Haematol 1998, 101(3): 539-47.
28. Antoku K, Liu Z, Johnson DE. Inhibition of caspase proteases by CrmA enhances the resistance of human leukemia cells to multiple chemotherapeutic agents. Leukemia 1997, 11(10): 1665-72.
29. Chen Z, Naito M, Mashima T, Tsuruo T. Activation of actin-cleavable interleukin 1 beta-converting enzyme (ICE) family protease CPP-32 during chemotherapeutic agent-induced apoptosis in ovarian carcinoma cells. Cancer Res 1996, 56(22): 5224-9.
30. Marissen WE, Lloyd RE. Eukariotic translation initiation factor 4G is targeted for proteolytic cleavage by caspase 3 during inhibition of translation in apoptotic cells. Mol Cell Biol 1998, 18(12): 7565-74.
31. Kraker AJ, Moore CW. Accumulation of cis-diamminedichloroplatinum (II) and platinum analogues by platinum-resistant murine leukemia cells in vitro. Cancer Res 1988; 48: 9-13.
32. Eichholtz-Wirth H, Hietel B. The relationship between cisplatin sensitivity and drug uptake into mammalian cells in vitro. Br J Cancer 1986; 54: 239-43.
33. Kawai K, Kamatani N, Georges E, Ling V. Identification of a membrane glycoprotein overexpression in murine lymphoma sublines resistant to cis- diamminedichloroplatinum (II). J Biol Chem 1990; 265: 13137-42.
34. Ishikawa T, Wright CD, Ishizuka H. GS-X pump Is functionally overexpressed in cis-diamminedichloroplatinum (II)-resistant human leukemia HL-60 Cells and downregulated by cell differentiation. J Biol Chem 1994; 269: 29085-93.
35. Goto S, Yoshido K, MoriKawa T, Urata Y, Suzuki K,. Kondo T. Augmentation of transport for cisplatin-glutathione adduct in cisplatin-resistant cancer cells. Cancer Res 1995; 55: 4297-301.
36. Veneroni S, Zaffaroni N, Daidone MG, Benini E, Villa R, Silvestrini R. Expression of P-glycoprotein and in vitro or in vivo resistance to doxorubicin and cisplatin in breast and ovarian cancers. Eur J Cancer 1994; 30A: 1002-7.
37. Jedlitschky G, Leier J, Buchholz U, Barnouin K, Kurz G, Keppler D. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res 1996; 56: 988-94.
38. Okuyama T, Maehara Y, Endo K, Baba H, Adachi Y, Kuwano M, Sugimachi K. Expression of glutathione S-transferase-pi and sensitivity of human gastric cancer cells to cisplatin. Cancer 1994; 74: 1230-6.
39. Awasthi S, Sharma R, Singhal SS, Herzog NK, Chaubey M, Awasthi YC. Modulation of cisplatin cytotoxicity by sulphasalazine. Br J Cancer 1994; 70: 190-4.
40. Bongers V, Snow GB, Braakhuis BJM. The role of glutathione S-transferases in head and neck squamous cell carcinogenesis. Eur J Cancer 1995; 31B: 349-54.
41 Minagawa Y, Kigawa J, Ishihara H, Itamochi H, Terakawa N. Synergistic enhancement of cisplatin cytotoxicity by SN-38, an active metabolite of CPT-11, for cisplatin-resistant HeLa cells. Jpn J Cancer Res 1994; 85: 966-71.
42. Boscia RE, Korbut T, Holden SA, Ara G, Teicher BA. Interaction of topoisomerase I inhibitors with radiation in cis-Diamminedichloroplatinum (II)-sensitive and -resistant cells in vitro and in the FSAIIC fibrosarcoma in vivo. Int J Cancer 1993; 53: 118-23.
43. Kikuchi Y, Hirata J, Yamamoto K, Ishi K, Kita T, Kudoh K, Tode T, Nagata I, Taniguchi K, Kuwano M. Altered expression of -glutamylcysteine synthetase, metallothionein and topoisomerase I or II during acquisition of drug resistance to cisplatin in human ovarian cancer cells. Jpn J Cancer Res 1997; 88: 213-7.
44. Hirata J, Kikuchi Y, Kita T, Imaizumi EE, Tode T, Ishii K, Kudoh K, Nagata I. Modulation of sensitivity of human ovarian cancer cells to cis-diamminedichloroplatinum (II) by 12-O-tetradecanoylphorbol-13-acetate and D,L-Buthionine-S,R-Sulphoximine. Int J Cancer 1993; 55: 521-7.
45.Sugimoto C, Matsukawa S, Fujieda S, Noda I, Tanaka N, Tsuzuki H, Saito H. Involvement of intracellular glutathione in induction of apoptosis by cisplatin in a human pharyngeal carcinoma cell line. Anticancer Res 1996; 16: 675-80.
46. Pendyala L, Perez R, Weinstein A, Zdanowicz J, Creaven PJ. Effect of glutathione depletion on the cytotoxicity of cisplatin and iproplatin in a human melanoma cell line. Cancer Chemother Pharmacol 1997, 40 (1): 38-44.
47. Kurokawa H, Nishio K, Ishida T, Arioka H, Fukuoka K, Nomoto T, Fukumoto H, Yokote H, Saijo N. Effect of glutathione depletion on cisplatin resistance in cancer cells transfected with the -glutamylcysteine synthetase gene. Jpn J Cancer Res 1997; 88: 108-10.
48. Campling BG, Baer K, Baker HM, Lam YM. Cole SPC. Do glutathione and related enzymes play a role in drug resistance in small cell lung cancer ctll lines? Br J Cancer 1993; 68: 327-35.
49. Kodera Y, Akiyama S, Isobe K, Kondo K, Ito K, Yamauchi M, Takagi H. Expression of -glutathione S-transferase gene (GSTP1) in gastric cancer: lack of correlation with resistance against cis-diamminedichloroplatinum (II). Eur J Cancer 1994; 30A: 2158-62.
50. Mellish KJ, Kelland LR, Harrap KR. In vitro platinum drug chemosensitivity of human cervical squamous cell carcinoma cell lines with intrinsic and acquired resistance to cisplatin. Br J Cancer 1993; 68: 240-50.
51. Eichholtz-Wirth H, Reidel G, Hietel B. Radiation-induced transient cisplatin resistance in murine fibrosarcoma cells associated with elevated metallothionein content. Br J Cancer 1993; 67: 1001-6.
52. Lazo JS, Basu A. Metallothionein expression and transient resistance to electrophilic antineoplastic drugs. Semin Cancer Biol 1991; 2: 267-71.
53. Hishikawa Y, Abe SI, Kinugasa S, Yoshimura H, Monden N. Overexpression of metallothionein correlates with chemoresistance to cisplatin and prognosis in esophageal cancer. Oncology 1997; 54: 342-7.
54. Vaisman A, Vachenko M, Said I, Chaney SG. Cell cycle changes associated with formation of Pt-DNA adducts in human ovarian carcinoma cells with different cisplatin sensitivity. Cytometry 1997; 27: 54-64.
55. Hill BT, Scanlon KJ, Hansson J, Harstrick A, Pera M, Fichtinger-Schepman AMJ, Shellard SA. Deficient repair of cisplatin-DNA adducts identified in human testicular teratoma cell lines established from tumours from untreated patients. Eur J Cancer 1994; 30A: 832-7.
56. Johnson SW, Swiggard PA, Handel LM, Godwin AK, Ozols RF, Hamilton TC. Relationship between platinum-DNA adduct formation and removal and cisplatin cytotoxicity in cisplatin-sensitive and resistant human ovarian cancer cells. Cancer Res 1994, 54: 5911-6.
57. Turchi JJ, Li M, Henkels KM. Cisplatin-DNA binding specificity of calf high mobility group I protein. Biochem 1996; 35: 2992-3000.
58.Ikeda S, Ozaki K. Action of mitochondrial endonuclease G on DNA damaged by L-ascorbic acid, peplomycin, and cis-diamminedichloroplatinum (II). Biochem Biophys Res Commun 1997, 235(2): 291-4.
59. Bissett D, McLaughlin K, Kelland LR, Brown R. Cisplatin-DNA damage recognition proteins in human tumor extracts. Br J Cancer 1993; 67:742-8.
60. McLaughlin K, Coren G, Masters J, Brown R. Binding activities of cis-platin-damage-recognition proteins in human tumor cell lines. Int J Cancer 1993; 53: 662-6.
61. Brown R, Hirst GL, Gallagher WM, McIlwrath AJ, Margison GP, Zee AG, Anthoney DA. HMLN1 expression and cellular responses of ovarian tumor cells to treatment with cytotoxic anticancer agents. Oncogene 1997, 15(1): 45-52.
62. Lanzi C, Perego P, Supino R, Romanelli S, Pensa T, Carenini N, Viano I, Colangelo D, Leone R, Apostoli P, Cassinelli G, Gambetta RA, Zunino F. Decreased drug accumulation and increased tolerance to DNA damage in tumor cells with a low level of cisplatin resistance. Biochem Pharmacol 1998, 55(8): 1247-54.
63. Fishel R, Ewel A, Lescoe MC. Purified human MSH2 protein binds to DNA containing mismatched nucleotides. Cancer Res 1994; 54: 5539-42.
64. Loeb LA. Microsatellite instability: marker of a mutator phenotype in cancer. Cancer Res 1994; 54: 5059-63.
65. Chu G. Cellular responces to Cisplatin. J Biol Chem 1994; 269: 787-90.
66. Sorenson CM, Eastman A. Influence of cis-Diamminedichloroplatinum (II) on DNA synthesis and cell cycle progression in excision repair proficient and deficient hamster ovary cells. Cancer Res 1988; 48: 6703-7.
67. Li Q, Gardner K, Zhang L, Tsang B, Bostick-Bruton F, Reed E. Cisplatin induction of ERCC-1 mRNA expression in A2780/CP70 human ovarian cancer cells. J Biol Chem 1998, 273 (36): 23419-25.
68. Husain A, He G, Venkatraman ES, Spriggs DR. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum (II). Cancer Res 1998, 58 (6): 1120-3.
69. Mestdach N, Pommery N, Saucier JM, Hecquet B, Founier C, Slomianny C, Teissier E, Henichart JP. Chemoresistance to doxorubicin and cisplatin in a murine cell line. Analysis of P-Glycoprotein, topoisomerase II activity, glutathione and related enzymes. Anticancer Res 1994; 14: 869-74.
70. Kondo H, Kanzawa F, Nishio K, Saito S, Saijo N. In vitro and in vivo effects of cisplatin and etoposide in combination on small cell lung cancer cell lines. Jpn J Cancer Res 1994; 85: 1050-6.
71. Jong S, Timmer-Bosscha H, Vries EGE, Mulder NH. Effect of novobiocin on cisplatin cytotoxicity and DNA interstrand cross-link formation in a cisplatin-resistant, small-cell lung carcinoma cell line. Int J Cancer 1993; 53: 110-7.
72. Guchelaar H-J, Timmer-Bosscha H, Dam-Meiring A, Uges DRA, Oosterhuis JW, Vries EGE Mulder NH. Enhancement of cisplatin and etoposide cytotoxicity after all-trans retinoic-acid-induced cellular differentiation of a murine embryonal carcinoma cell line. Int J Cancer 1993; 55: 442-7.
73. Seki S, Hongo A, Zhang B, Akiyama K, Sarker AH, Kudo T. Inhibition of cisplatin-mediated DNA damage in vitro by ribonucleotides. Jpn J Cancer Res 1993; 84: 462-7.
74. Parekh HK, Simpkins H. The differential expression of citokeratin 18 in cisplatin-sensitive and -resistant human ovarian adenocarcinoma cells and its association with drug sensitivity. Cancer Res 1995; 55: 5203-6.
75. Mirakhur B, Parekh HK, Simpkins H. Expression of the cisplatin resistance phenotype in a human ovarian carcinoma cell line segregates with chromosomes 11 and 16. Cancer Res 1996; 56: 2256-62.
76. Mueller H, Eppeberger U. The dual role of mutant p 53 protein in chemosensitivity of human cancers. Anticancer Res 1996, 16 (6B): 3845-8.
77. Kawasaki t, Tomita Y, Bilim V, Takeda M, Takahashi K, Kumanishi T. Abrogation of apoptosis induced by DNA-damaging agents in human bladder-cancer cell lines with p 21/WAF1/CIP1 and/or p 53 gene alterations. Int J Cancer 1996, 68 (4): 501-5.
78. Kondo S, Barna BP, Kondo Y, Tanaka Y, Casey G, Liu J, Morimura T, Kaakaji R, Peterson JW, Werbel B, Barnett GH. WAF1/CIP1 increases the susceptibility of p 53 non-functional malignant glioma cells to cisplatin-induced apoptosis. Oncogene 1996, 13 (6): 1279-85.
79. Li G, Tang L, Zhou X, Tron V, Ho V. Chemotherapy-induced apoptosis in melanoma cells is p 53 dependent. Melanoma Res 1998, 8 (1): 17-23.
80. Biagosklonny MV, El-Deiry WS. Acute overexpression of wt p 53 facilitates anticancer drug-induced death of cancer and normal cells. Int J Cancer 1998, 75 (6): 933-940.
81. Li R, Sutphin PD, Schwartz D, Matas D, Almog N, Wolkowicz R, Goldginger N, Pei H, Prokocimer M, Rotter V. Mutant p 53 protein interferes with p 53-independent apoptotic pathways. Oncogene 1998, 16 (25): 3269-77.
82. Vasey PA, Jones NA, Jenkins S, Dive C, Broun R. Cisplatin, camptothecin, and taxol sensitivities of cells with p 53-associated multidrug resistance. Mol Pharmacol 1996, 50 (6): 1536-40.
83. Vikhanskaya F, Clerico L, Valenti M, Stazione NS, Broggini M, Parodi S, Russo P. Mechanism of resistance to cisplatin in a human ovarian-carcinoma cell line selected for resistance to doxorubicin: possible role of p 53. Int J Cancer 1997, 72 (1): 155-9.
84. Vaisman A, Varchenko M, Said I, Chaney SG. Cell cycle changes associated with formation of Pt-DNA adducts in human ovarian carcinoma cells with different cisplatin sensitivity. Cytometry 1997, 27 (1): 54-64.
85. Siddik ZH, Mims B, Lozano G, Thai G. Independent pathways of p 53 induction by cisplatin and X-rays in a cisplatin-resistant ovarian tumor cell line. Cancer Res 1998, 58 (4): 698-703.
86. Ju JF, Banerjee D, Lenz HJ, Danenberg KD, Schmittgen TC, Spreas CP, Schonthal AH, Manno DJ, Hochhauser D, Bertino JR, Danenberg PV. Restoration of wild-type p 53 activity in p 53-null HL-60 cells confers multidrug sensitivity. Clin Cancer Res 1998, 4 (5): 1315-22.
87. Fajac A, Da Silva J, Ahomadegbe JC, Rateau JG, Bernaudin JF, Riou G, Benard J. Cisplatin-induced apoptosis and p 53 gene status in a cisplatin-resistant human ovarian carcinoma cell line. Int J Cancer 1996, 86 (1): 67-74.
88. Kondo S, Barna BP, Morimura T, Takeuchi J, Yuan J, Akbasak A, Barnett GH. Interleukin-1-beta-converting enzyme mediates cisplatin-induced apoptosis in malignant glioma cells. Cancer Res 1995, 55 (24): 6166-71.
89. De Feudis P, Debernardis D, Beccaglia P, Valenti M, Sire GE, Arzani D, Stanzione S, Parodi S, D`Incalci M, Russo P, Broggini M. DDP-induced cytotoxicity is not influenced by p 53 in nine human ovarian cancer cell lines with different p 53 status. Br J Cancer 1997, 76 (4): 474-9.
90. Burger H, Nooter K, Boersma AW, Kortland CJ, Stoter G. Expression of p 53, Bcl-2 and Bax in cisplatin-induced apoptosis in testicular germ cell tumor cell lines. Br J Cancer 1998, 77 (10): 1562-7.
91. Ikeguchi M, Tatebe S, Kaibara N, Ito H. Changes in levels of expression of p 53 and the product of the bcl-2 in lines of gastric cancer cells during cisplatin-induced apoptosis. Eur Surg Res 1997, 29 (5), 396-402.
92. Pestell KE, Medlow CJ, Titley JC, Kelland LR, Walton MI. Characterisation of the p 53 status, BCL-2 expression and radiation and platinum drug sensitivity of a panel of human ovarian cancer cell lines. Int J Cancer 1998, 77 (6): 913-8.
93. Nakata B, Chung KH, Ogawa M, Yanagawa K, Muguruma K, Inoue T, Yamashita Y, Onoda N, Maeda K, Sawada T, Sowa. P 53 protein overexpression as a predictor of the response to themotherapy in gastric cancer. Surg Today 1998, 28 (6): 595-8.
94. Petty R, Evans A, Duncan I, Kurbacher C, Cree I. Drug resistance in ovarian cancer – the role of p 53. Payhol Oncol Res 1998, 4 (2): 97-102.
95. Simonian PL, Grillot DA, Andrews DW, Leber B, Nunez G. Bax homodimerization is not required for Bax to accelarate chemotherapy-induced cell death. J Biol Chem 1996, 271 (50): 32073-7.
96. Liu JR, Fletcher B, Page C, Hu C, Nunez G Baker V. Bcl-xL is expressed in ovarian carcinoma and modulates chemotherapy-induced apoptosis. Gynecol Oncol 1998, 70 (3), 398-403.
97. Miyake H. Hanada N, Nakamura H, Kagawa S, Fujiwara T, Hara I, Eto H, Gohji K, Arakawa S, Kamidono S, Saya H. Overexpression of Bcl-2 in bladder cancer inhibits apoptosis induced by cisplatin and adenoviral-mediated p 53 gebe transfer. Oncogene 1998, 16 (7): 1933-43.
98. Minn AJ, Rudin CM, Boise LH, Thompson CB. Expression of bcl-xl can confer a multidrug resistance phenotype. Blood 1995, 86 (5): 1903-10.
99.Simonian PL, Grillot AM, Nunez G. Bcl-2 and Bcl-Xl can differentially block chemotherapy induced cell death. Blood 1997, 90 (3): 1208-16.
100. Jones NA, Turner J, McIlwrath AJ, Brown R, Dive C. Cisplatin- and paclitaxel-induced apoptosis of ovarian carcinoma cells and the relationship between bax and bak up-regulation and the functional status of p 53. Mol Pharmacol 1998, 53 (3): 819-26.
101. Boersma AW, Nooter K, Burger h, Kortland CJ, Stoter G. Bax upregulation is an early event in cisplatin-induced apoptosis in human testicular germ-cell tumor cell line NT2, as quantitated by flow cytometry. Cytometry 1997, 27 (3): 275-82.
102. Lee JU, Hosotani R, Wada H, Doi R, Kosiba T, Fujimoto K, Miyamoto Y, Mori C, Nakamura N, Shiota K, Imamura M. Mechanism of apoptosis induced by cisplatin and VP-16 in PANC-1 cells. Anticancer Res 1997, 17 (5A): 3445-50.
103. Houldsworth J, Xiao H, Murty VV, Chen W, Ray B, Reuter VE, Bosl GJ, Chaganti RS. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16 (18): 2345-9.
104. Sakahura C, Sweeney EA, Shirahama T, Igarashi Y, Hakomori S, Tsujimoto H, Imanishi T, Ogaki M, Yamazaki J, Hagiwara A, Yamaguchi T, Sawai K, Takahashi T. Overexpression of bax sensitizes breast cancer MCF-7 cells to cisplatin and etoposide. Surg Today 1997, 27 (7): 676-9.
105. Kondo S, Shinomura Y, Kanayama S, Higashimoto Y, Kiyohara T, Zushi S, Kitamura S, Ueyama H, Matsuzawa Y. Modulation of apoptosis by endogenous Bcl-Xl expression in MKN-45 human gastric cancer cells. Oncogene 1998, 17(20): 2585-91.
106. Nakata B, Muguruma K, Hirakawa K, Chung YS, Yamashita Y, Inoue T, Matsuoka T, Onoda N, Kato Y, SowaM. Predictive value of Bcl-2 and Bax protein expression for chemotherapeutic effect in gastric cancer. Apilot study. Oncol 1998, 55(6): 543-7.
107. Boku N, Chin K, Hosokawa K, Ohtsu A, Tajiri H, Yoshida S, Yamao T, Kondo H, Shirao K, Shimada Y, Saito D, Hasebe T, Mukai K, Seki S, Saito H, Johnston PG. Biological markers as a predictor for response and prognosis of unresectable gastric cancer patients treated with 5-fluorouracil and cis-platinum. Clin Cancer Res 1998, 4 (6): 1469-74.
108. Herod JJ, Eliopoulos AG, Warwick J, Niedobitek G, Young LS, Kerr DJ. The prognostic significance of bcl-2 and p 53 expression in ovarian carcinoma. Cancer Res 1996, 56 (9): 2178-84.
109. Muguruma K, Nakata B, Hirakawa K, Yamashita Y, Onoda N, Inoue T, Matsuoka T, Kato Y, Sowa. P 53 and Bax protein expression as predictor of chemotherapeutic effect in gastric carcinoma. Gan To Kagaku Ryoho 1998, 3: 400-3.
110. Ferguson AW, Flatow U, MacDonald NJ, Larminat F, Bohr VA, Steeg PS. Increased sensitivity to cisplatin by nm23-transfected tumor cell lines. Cancer Res 1996; 56: 2931-5.
111.Masumoto N, Nakaano S. Cell signalling and CDDP resistance. Gan To Kagaku Ryoho 1997, 24(4): 424-30.
112. Demidova NS, Ilynskaya GV, Shiryaeva OA, Chernova OB, Goncharova SA, Kopnin BP. Decreased sensitivity of multidrug-resistant tumor cells to cisplatin is correlated with sorcin gene co-amplification. Neoplasma 1995; 42: 195-201.
Похожие работы
... правило, выражен асцит. Ректовагинальное исследование необходимо для определения инвазии ракового процесса в параректальную и параметральную клетчатку. Современная диагностика злокачественных опухолей яичников включает в себя трансвагинальную эхографию с применением акустических излучателей, обладающих высокой разрешающей способностью при непосредственном соприкосновении сканирующей поверхности ...
... произошли изменения не менее чем в 6 – 10 генетических факторах (теория многоступенчатого канцерогенеза). Изменения в пределах всего одной копии или аллеля протоонкогена достаточны для превращения его в онкоген с возросшей стимулирующей пролиферацию активностью, т.е. онкогены можно рассматривать как доминантные трансформирующие гены. Антионкогены, напротив, проявляются рецессивным образом т.е. ...
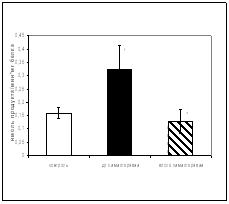
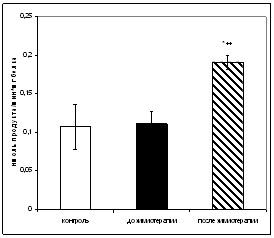
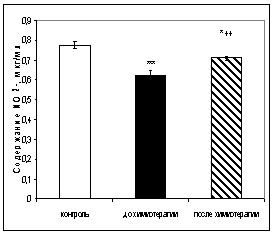
... больных до и после химиотерапии может быть использовано в качестве дополнительных биохимических критериев при наблюдении за эффективностью проводимого лечения [6,33]. 3.2 Исследование активности ангиотензинпревращающего фермента в сыворотке крови онкологических больных при химиотерапевтическом воздействии Результаты исследования показали, что активность АПФ у онкологических больных до ...
... на функцию легких, мочеполовую систему, кожи и ее придатки, гипертермические реакции, токсические флебиты и других проводится в зависимости от характера осложнений. Непосредственные особенности анестезиологического пособия Резекция легких. Опухоли легких могут быть доброкачественнми, злокачественными или занимать промежуточное положение. Лишь в редких случаях удается составить мнение о ...
0 комментариев