Курсовая работа
ВведениеСущность физико-химических методов анализа заключается в том, что на основании измерения величины, характеризующей какое-нибудь свойство раствора, определяют концентрацию в нем исследуемого компонента. Исследованию могут быть подвергнуты не только жидкие, но и твердые растворы (например, металлические сплавы) или газовые смеси. Обычно между количественной характеристикой исследуемого свойства и концентрацией анализируемого вещества существует определенная количественная зависимость, которая может быть выражена при помощи так называемого уравнения связи. В большинстве случаев уравнение связи имеет простую форму:
P=εC
где Р—количественная характеристика исследуемого свойства;
ε—постоянная (коэффициент пропорциональности);
С— концентрация.
Уравнениями такого типа выражается, например, зависимость оптической плотности от концентрации окрашенного вещества (закон Бугера—Ламберта—Бера), силы диффузионного тока от концентрации электролита (уравнение Ильковича), электропроводности от концентрации и т. д.
Лишь в сравнительно редких случаях уравнение связи имеет другую форму. Так, например, величина электродвижущей силы концентрационного элемента находится в линейной зависимости от логарифма концентрации исследуемого иона.
Чувствительность данного физико-химического метода определяется возможностью измерения минимальных значений величины Р и величиной коэффициента пропорциональности ε.
Скорость химической реакции зависит от ряда факторов, в том числе от концентрации реагирующих веществ (в частном случае от концентрации катализаторов). Эта зависимость может быть использована в аналитических целях.
1. Кинетические методы анализаМетоды анализа, основанные на измерении скорости реакции и использовании ее величины для определения концентрации, объединяются под общим названием кинетических методов анализа. В литературе можно найти описание различных вариантов кинетических методов анализа, которые в зависимости от типа используемых реакций или способа измерения различных кинетических характеристик называются каталиметрическими, хронометрическими, темпометрическими и др.
Кинетические методы анализа могут применяться как для определения сравнительно больших концентраций, так и для определения очень малых концентраций различных веществ.
В первом случае, как правило, используют обычные реакции, во втором — каталитические. Использование некаталитических реакций и определение средних концентраций при помощи кинетических методов представляет интерес преимущественно для органической химии. Каталитические реакции особенно важны для определения очень малых концентрации различных ионов в неорганическом анализе, так как они характеризуются исключительно высокой чувствительностью, примерно равной чувствительности активационного анализа и превосходящей чувствительность спектрального и спектрофотометрического методов анализа. Чувствительность последних двух методов почти никогда не превосходит сотых долей микрограмма в миллилитре. При помощи каталитических реакций можно определить тысячные, десятитысячные и даже миллионные доли микрограмма в миллилитре. Например, золото и марганец при помощи каталитических реакций определяют при концентрации их порядка 0,00001 мкг/мл, а кобальт даже при концентрации 0.000001 мкг/мл.
Чувствительность любого физико-химического метода анализа, как видно из уравнения, определяется отношением P/ε и, следовательно, зависит от возможностей измерить минимальную количественную характеристику данного свойства (Р) и от величины коэффициента ε. Измерению доступны крайне небольшие скорости реакций, если выбранный для наблюдения отрезок времени достаточно велик, а метод измерения меняющейся концентрации достаточно чувствителен. Величина коэффициента ε при использовании каталитических реакций весьма велика.
Это объясняется тем, что за сравнительно небольшой отрезок времени частица катализатора участвует во многих элементарных актах и, таким образом, одна частица катализатора вызывает появление огромного числа (миллионов, миллиардов) частиц продуктов реакции, открываемых различными способами. В этом отношении можно провести известную аналогию между явлениями, протекающими при каталитических реакциях, и явлениями, наблюдаемыми в камере Вильсона или в газовом счетчике. Во всех рассматриваемых случаях одна частица вызывает превращения (химические изменения, ионизацию) многих частиц. В случае каталитических реакций в отличие от процессов, наблюдаемых в камере Вильсона, число вновь появляющихся в результате действия катализатора частиц неограниченно и зависит только от длительности наблюдения.
Обычно определения при помощи кинетических методов выполняются довольно быстро и сравнительно просто, без применения дорогостоящих или очень сложных приборов. Следует отметить, что применение специальных устройств для термостатирования реакционных сосудов и автоматизации записи процесса, протекающего в реакционной смеси, позволяет повысить точность метода. Повышение же чувствительности измерения концентрации с применением специальных устройств дает возможность увеличить чувствительность метода.
Кинетические методы анализа отличаются универсальностью: индикаторные каталитические реакции предложены для определения около 40 элементов периодической системы Д. И. Менделеева. Такие реакции неизвестны пока лишь для я элементов первых трех главных групп. Однако для этих элементов известен ряд микробиологических реакций и биологических тестов, поэтому можно полагать, что и для них будут найдены соответствующие каталитические реакции.
В некоторых случаях кинетические методы характеризуются исключительной специфичностью. Можно назвать, например, реакцию окисления тиосульфат-иона соединениями железа (III). Эта реакция катализируется только соединениями меди. Особенно резко повышается специфичность реакции, если катализатором является комплексное соединение металла.
Специфичность каталитических реакций может быть повышена путем введения в анализируемый раствор веществ, маскирующих каталитически активные примеси. При введении в раствор маскирующие агенты с примесями образуют неактивные комплексы. Особенно интересен случай добавления к раствору вещества, являющегося активатором для определяемого элемента, и маскирующим агентом для примесей.
Скорость химической реакции сильно зависит от температуры, наличия примесей в растворе, ионной силы, а иногда от величины и состояния стенок реакционного сосуда. Поэтому при выполнении измерений необходимо очень тщательно термостатировать реакционные сосуды, применять исключительно чистые растворители и реактивы и соблюдать возможно большее сходство в условиях проведения химической реакции и исследуемом растворе и в растворе сравнения. При современном уровне техники выполнения кинетических исследований не всегда удается достаточно строго унифицировать все эти условия, поэтому воспроизводимость кинетических методов анализа обычно характеризуется средним отклонением порядка нескольких процентов (но не более 10%).
Однако воспроизводимость определения можно резко увеличить, если каталитические реакции использовать для определения точки эквивалентности при титровании катализаторов ингибиторами или наоборот. Такой метод объемного анализа назван каталиметрическим титрованием. Он дает возможность при титровании весьма разбавленных растворов достигнуть довольно высокой точности и воспроизводимости (десятые доли процента).
Кинетические методы анализа представляют наибольший интерес для определения очень малых концентраций элементов. Эти методы могут быть использованы при определении содержания примесей в полупроводниковых материалах. С успехом эти методы применяют для определения содержания микроэлементов в биологических объектах, при анализе металлов и сплавов, горных пород, грунтовых вод, а также при анализе реактивов и материалов особо высокой чистоты.
Как правило, при использовании кинетических методов анализа определяют равновесную, а не общую концентрацию вещества, поэтому указанные методы с успехом можно применять при изучении равновесий в растворах и, в особенности, при изучении равновесий реакций образования комплексных соединений.
1.1. Методы измерения скорости реакцииДля изучения кинетики реакции и определения ее скорости необходимо знать изменение во времени концентрации хотя бы одного из реагирующих веществ или продуктов реакции.
Для определения концентрации веществ можно применять как химические, так и физико-химические методы анализа.
Химические методы анализа можно применять для изучения медленно протекающих реакций. При изучении реакций, протекающих со значительными скоростями, необходимо быстро останавливать их течение одним из следующих способов:
1) резким охлаждением реакционной смеси, для чего, например, быстро смешивают реакционную смесь с сильно охлажденным растворителем;
2) добавлением ингибитора, образующего прочное соединение с катализатором;
3) добавлением вещества, которое мгновенно связывает одно из реагирующих веществ;
4) резким изменением рН раствора (добавлением кислоты или основания).
После того как реакция остановлена, анализируют реакционную смесь.
Из химических методов объемные определения являются наиболее распространенными. На пример, изучение кинетики разложения перекиси водорода в щелочных растворах в присутствии катализаторов. Эта реакция катализируется очень малыми примесями солей меди; скорость ее контролируют отбором проб через известные промежутки времени и определением содержания в них перекиси водорода иодометрическим методом (добавляют иодид калия, молибдат аммония, подкисляют и титруют выделившийся иод тиосульфатом).
Главное достоинство химических методов анализа — возможность непосредственного измерения абсолютных концентраций реагирующих веществ или продуктов реакции. Недостатки этих методов—невозможность непрерывного измерения концентрации и, как правило, продолжительность самого определения.
В физико-химических методах анализа измеряется изменение во времени какого-нибудь физического свойства системы (раствора). Например, изменение объема выделяющегося газа, оптической плотности раствора, показателя преломления, электропроводности, силы диффузионного тока, потенциала определенного электрода, люминесценции и т. п.
Достоинствами этой группы методов является быстрота определений, возможность выполнения измерений непосредственно в реакционном сосуде без предшествующего отбора проб, без нарушения равновесия в системе. Во многих случаях удается проводить непрерывную и даже автоматическую запись, благодаря чему в распоряжении экспериментатора имеется по сути неограниченное число точек.
Подавляющее большинство физических свойств веществ в растворах связано линейной зависимостью с их концентрацией:
Р= εхχ
где Р—количественная характеристика свойства раствора;
χ—концентрация продукта реакции X;
εх—коэффициент пропорциональности (например, молярный коэффициент погашения вещества X).
Если данное свойство характерно для нескольких веществ (например, для А и X), то зависимость более сложная:
Р=εa(a-χ)+ εхχ
После введения обозначений Р0= εaa и P∞= εaa и преобразований получаем:
Р=P0+( P∞-P0)x/a
Газоволюметрический метод определения скорости реакции основан на измерении объема выделяющегося газообразного продукта реакции. Между объемом газа и числом молей образовавшегося продукта реакции существует простейшая зависимость, определяемая законом Авогадро.
Для стандартных условий (температура 25 °С и давление 1 атм) число молей (п) образовавшегося продукта реакции
равно:
n=V/24400
где V—объем выделившегося газа, мл.
В качестве примера можно привести изучение кинетики каталитического разложения перекиси водорода в щелочном растворе на основании, измерения объема выделившегося кислорода нитрометром Лунге.
Из оптических методов при изучении кинетики каталитических реакций в растворах наибольшее распространение в последние годы получили колориметрический и спектрофотометрический методы анализа. Оба метода основаны на измерении оптической плотности растворов и отличаются лишь тем, что в случае колориметрического метода используют белый свет или участки спектра, выделенные при помощи широкополосых светофильтров, а в случае спектрофотометрического анализа применяется монохроматический свет.
Между оптической плотностью и концентрацией окрашенного вещества в растворе существует простая зависимость, вытекающая из закона Бугера—Ламберта—Бера:
D=εlC
где D—оптическая плотность раствора;
ε — молярный коэффициент погашения;
l—толщина поглощающего слоя.
В качестве примеров применения этих методов приведем каталитические реакции окисления иодид-иона перекисью водорода и тиосульфат-иона—ионом железа (III)
H2O2 + 2I- + 2H+ = 2H2O + I2
2Fe3+ + 2S2O32- = 2Fe2+ + S4O62-
В первом случае в реакционную смесь вводят крахмал и скорость реакции измеряют по оптической плотности образовавшегося иодкрахмала (оптическая плотность непрерывно увеличивается), во втором случае в раствор добавляют роданид калия или аммония и скорость реакции измеряют по оптической плотности роданидного комплекса железа (оптическая плотность непрерывно уменьшается).
Для изучения кинетики реакции указанными методами используют любой прибор, на котором возможно измерение оптической плотности (фотометры самых различных типов, любые фотоэлектроколориметры, спектрофотометры).
Чувствительность оптических методов анализа может быть значительно повышена, если в качестве рецептора применить фотоумножители вместо обычных фотоэлементов.
Для измерения скорости реакции можно применять также турбидиметрические и нефелометрические методы. В качестве примера приведем реакцию окисления тиосульфат-иона перекисью водорода с образованием сульфат-иона:
4Н2O2 + S2O32- = 3H2O + 2SO42- + 2H+
В исследуемый раствор вводят хлорид бария и желатин. Образующийся при этой реакции сульфат-ион дает суспензию BaSO4, оптическая плотность которой может быть измерена на фотоэлектроколориметре. Но так как оптическая плотность суспензии пропорциональна концентрации сульфат-иона в растворе, то все приведенные выше рассуждения применимы и к этому случаю.
Применение люминесцентного метода определения скорости химической реакции весьма перспективно, так как вследствие большой чувствительности метода по изменению люминесценции раствора можно определять очень малые изменения концентрации реагентов в растворе. В качестве индикаторной была выбрана реакция окисления флуоресцирующего вещества — стильбексона перекисью водорода. Эта реакция катализируется железом.
О скорости реакции можно судить по изменению величины lgI0/I во времени (I0 и I - интенсивность флуоресценции в начальный и в данный момент времени, соответственно).
При каталитических реакциях, сопровождающихся хемилюминесценцией, скорость реакции также может быть измерена методом люминесцентного анализа. Так, например, А. К. Бабко и Н. М. Луковская при изучении реакции между перекисью водорода и люминолом в присутствии солей меди применяли фотографический метод оценки интенсивности свечения. При действии выделяющегося при хемилюминесценции света на фотопластинку происходит ее почернение (после проявления и фиксирования). Интенсивность почернения зависит от концентрации реагирующих веществ.
Применение потенциометрического метода измерения скорости химических реакций изучено на реакции окисления тиосульфат-ионов ионами железа (III), которая катализируется солями меди.
В этом случае можно применять платиновый электрод в качестве индикаторного и для измерения скорости реакции использовать концентрационную цепь.
При полярографическом методе измерения скоростей химических реакций используют зависимость между силой диффузионного тока и концентрацией определяемого вещества. Так, за изменением концентрации хромовой кислоты в процессе ее восстановления можно следить по величине диффузионного полярографического тока.
Даже небольшие изменения в соотношении концентраций иодида и иода в растворе можно регистрировать амперометрически при помощи двух платиновых индикаторных электродов. Сила тока в системе зависит от концентрации иода, выделяющегося при окислении иодида.
Такую систему, следовательно, можно использовать для измерения скорости выделения иода из иодида при окислении его, например, перекисью водорода. Скорость этой реакции, как известно, зависит от концентрации катализаторов (молибдена, вольфрама и др.) в растворе. При протекании реакции в растворе сила тока линейно изменяется во времени. По тангенсу угла наклона прямой время—сила тока можно найти скорость соответствующей реакции и определить концентрацию элемента-катализатора в растворе.
2. Термодинамический вывод диаграммы состоянияПри исследовании систем, состоящих из двух или большего числа химических индивидов, главную роль играет зависимость свойств системы от cостава. Измеряется то или иное свойство для смесей или растворов различного состава, по возможности от 0 до 100 % каждого из исходных индивидов, и строится диаграмма состав - свойство или эта зависимость дается аналитически. Несмотря на то, что последний способ представления результатов является более высокой ступенью в обработке результатов измерения, в физико-химическом анализе пока используется преимущественно графический метод. Геометрический образ — диаграмма — отражает, какие процессы прошли в системе: образовались ли механические смеси, твердые или жидкие растворы, возникли ли новые соединения и т. д. По диаграмме также определяются границы существования различных фаз в системе. Анализ диаграммы позволяет выявить не столь резко выраженные процессы и отметить слабые межчастичные взаимодействия, которые не приводят к образованию новых соединений или распаду имеющихся.
Естественно, в одном реферате рассмотреть все диаграммы состояния для всех систем просто невозможно. Поэтому, в качестве примера мы решили рассмотреть диаграммы состояния двойных конденсированных систем без превращений в твердых фазах.
2.1. Термодинамический вывод диаграммы состояния системы с простой эвтектикойРассмотрим изотермы удельных изобарных потенциалов расплавов двойной системы В—А для разных температур. Установим, какие фазы находятся в равновесии при той или иной температуре, и построим диаграмму зависимости температур от состава системы, т. е. диаграмму состояния.
На рис.I изображены изотермические диаграммы удельного изобарного потенциала этой системы в жидком состоянии для температур t1 › t2 › t3 › t4 › t5 › t6 (диаграммы I—VI; VII — полученная из них диаграмма состояния). Точками Аs и Вs на всех диаграммах обозначены удельные изобарные потенциалы компонентов в твердом состоянии. Точки Al и Bl (концевые точки кривых) относятся к тем же компонентам в жидком состоянии.
Пусть температура t1 выше температуры плавления более высокоплавкого компонента А. При этой температуре точки Аs и Вs лежат соответственно выше точек Al и Bl (диаграмма I). Следовательно, смеси всех составов при температуре t1 будут в жидком состояни. При понижении температуры изобарный потенциал возрастает, так как dG/dT =-S, где S — энтропия, величина всегда положительная. Энтропия одного и того же вещества в жидком состоянии больше, чем в твердом при той же температуре, поэтому с понижением температуры изобарный потенциал жидкости возрастает быстрее, чем твердого тела. Вследствие этого при более низких температурах устойчиво твердое вещество. Эти различия в температурном ходе изобарного потенциала твердых и жидких веществ обусловливают и вывод диаграммы с начала кристаллизации расплавов, для которого важно относительное движение точек Аs (Вs) и Al (Bl). При понижении температуры точка Аs, сближается с точкой Al, а точка Вs — с Bl, и при температуре плавления компонента А (t2) точки Аs и Al совпадают, точка же Вs лежит пока еще выше Bl, и при температуре t2 компонент В находится в расплавленном состоянии (диаграмма II).
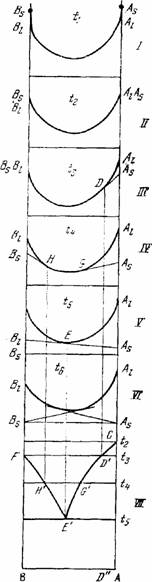
Рис.1. Вывод диаграммы состояния двойной системы с простой эвтектикой из изотермы изобарного потенциала.
Диаграмма III соответствует t3 - температуре плавления компонента В; точки Вs, и Bl; совпадают. Точка Аs находится ниже Al. Проведем из точки Аs касательную к кривой и через точку касания D — вертикальную линию. Отметим на диаграмме состояния (диаграмма VII) температуру t3 и проведем соответствующую ей изотермическую прямую F t3. Точка пересечения прямой F t3 с указанной выше вертикальной прямой (точка D') указывает температуру начала затвердевания системы с составом, изображаемым точкой D. Кроме того, отметим на диаграмме точки плавления компонентов А и В (точки С и F).
При дальнейшем понижении температуры до t4 (диаграмма IV), которая немного ниже температуры плавления более низкоплавкого компонента В, обе точки Аs и Вs, по сравнению с Al и Bl окажутся еще ниже, чем при температуре t3. Из этих точек можно провести касательные к кривой, причем точки касания G и Н дают составы растворов, находящихся при этой температуре в равновесии с твердым А и соответственно с твердым В. Точка G, как легко видеть, будет лежать левее, чем точка D диаграммы VII. Пользуясь диаграммой VII, получим соответствующие точки Н' и С' на диаграмме состояния при помощи описанного выше приема. При продолжающемся падении температуры точки Аs и Вs будут претерпевать дальнейшее понижение относительно Al и Bl, вследствие чего точки касания G и Н будут двигаться навстречу друг другу (первая налево, а вторая направо). Наконец, при температуре t5 (диаграмма V), когда обе точки касания встретятся в точке Е, обе касательные сольются в одну прямую. Это означает, что при этой температуре оба твердых компонента находятся в равновесии с одним и тем же раствором, состав которого дается точкой Е. Перенесем уже описанным способом точку Е на диаграмму состояния и получим точку Е'.
Если соединить точки, полученные указанным выше способом на диаграммеме VII, то получим две линии температур начала затвердевания: FН'Е’, отвечающую выделению компонента В, и CD’G'Е', отвечающую выделению компонента А. Таким образом, кривая температур начала кристаллизации диаграммы состояния окажется построенной. Эта кривая называется ликвидусом и состоит из двух ветвей, соответствующих кристаллизации того и другого компонента. Ветви пересекаются в точке Е', которая будет изображать состояние раствора (расплава), находящегося в равновесии одновременно с твердыми В и А. Раствор, находящийся в равновесии с двумя твердыми фазами, называется двояконасыщенным. При продолжающемся отнятии теплоты от системы температура и состав жидкости, состояние которой определяется течкой Е', постоянны. Расплав Е' называется эвтектическим или жидкой эвтектикой. Затвердевшая жидкая эвтектика называется твердой эвтектикой ( по валовому составу они тождественны), а температура, при которой такая эвтектика затвердевает,— эвтектической температурой. Точка Е' изображающая состояние жидкой эвтектики (фигуративная точка жидкой эвтектики), называется эвтектической точкой. Когда это не может повести к недоразумению, употребляют один термин — эвтектика, объединяя и температуру, и состав эвтектической точки. Так как в эвтектике двойной системы число компонентов равно двум, число фаз — трем, а давление постоянно, то эта точка нонвариантная (точнее, условно-нонвариантная).
Вернемся к рис.1. При дальнейшем понижении температуры (диаграмма VI) точки Аs и Вs и соединяющая их прямая еще более опустятся по отношению к кривой. Из этих точек можно, конечно, провести касательные к кривой и определить, таким образом, растворы, находящиеся в равновесии с твердыми А и В. Однако эти равновесия отвечают неустойчивым состояниям, что соответствует возможности продолжить кривую плавкости за эвтектическую точку. В устойчивых же состояниях будут находиться механические смеси твердых А и В, так как при данном составе удельный изобарный потенциал смеси будет меньше, чем смеси твердых А или В с находящимися с ними в равновесии растворами.
Список литературыК.Б. Яцимирский ″Кинетические методы анализа″, М.:″ Химия″ 1967г
В.Я. Аносов, М.И. Озерова, Ю.Я. Фиалков ″Основы физико-химического анализа″, М.: ″Наука″,1
Похожие работы
... коэффициент конденсации [13]. Более общая задача, описывающая диффузионный рост системы зародышей при наличии нестационарного реиспарения адатомов металла, рассмотрена в [14, 15]. При экспериментальном исследовании физико-химических закономерностей энергообмена установлено, что между энергией реиспаренных атомов W и физико-химическими свойствами материала подложки наблюдается корреляция [10, ...

... основные закономерностей активации LiAl, LixC6 и С8С3 электродов путем механических, физико-химических и электрохимических воздействий, а также изучние обратимой работа модифицированных электродов, работающих по "принципу электрохимического внедрения, в макетах литиевых аккумуляторов. Задачи исследования. Для достижения поставленной цели потребовалось: -провести комплексное систематическое ...
... хлоридом аммония в вакууме по методике, разработанной авторами [ 98, 108 ]. Глава III Исследование механизма электровосстановления ионов самария в хлоридных и хлоридно - фторидных расплавах. Исследование процесса электровосстановления ионов Sm3+ вольтамперометрическим методом при стационарных и нестационарных режимах поляризации ...

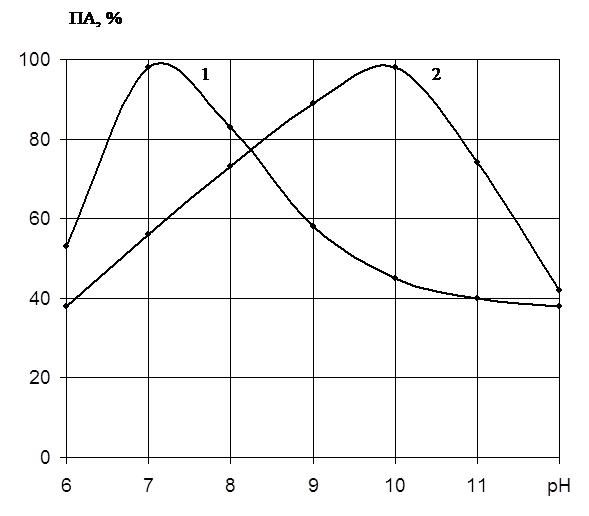
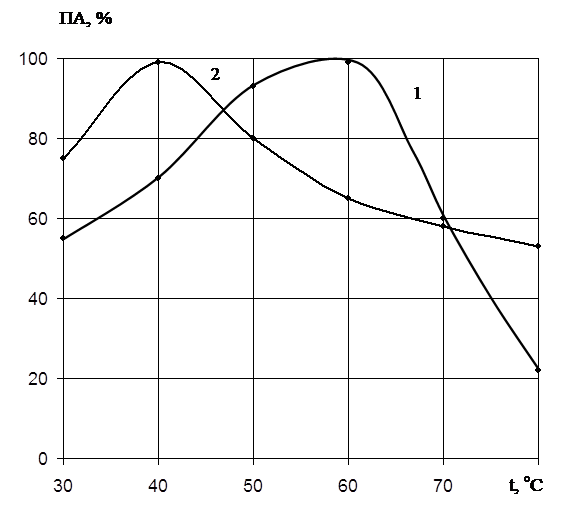
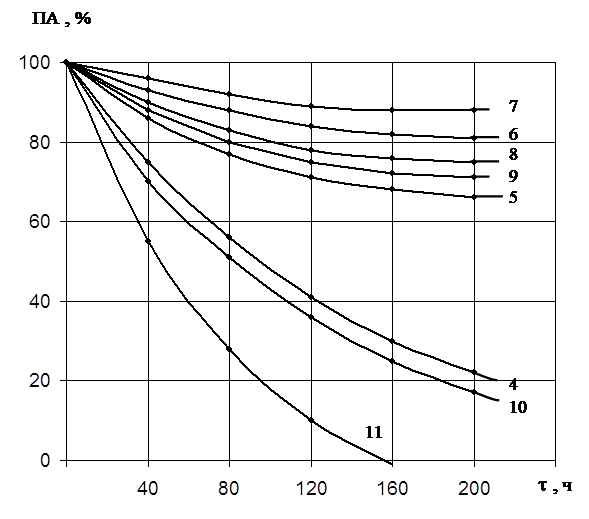
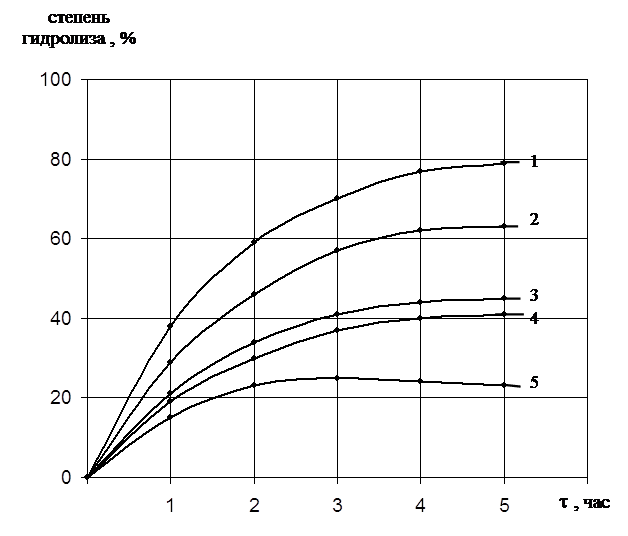
... есть среднее из двух или трех определений. Обсуждаются только те результаты, которые были воспроизводимы в каждом опыте. ГЛАВА 3. ПОЛУЧЕНИЕ ПРЕПАРАТОВ ПРОТЕИНАЗЫ PENICILLIUM WORTMANNII 2091 И ИССЛЕДОВАНИЕ ИХ ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ. Известно, что микроорганизмы синтезируют богатые набором ферментов комплексы. Поэтому важным этапом в получении препаратов направленного действия является ...
0 комментариев