Украинский Государственный
Химико-Технологический Университет
Заочный факультет
Дисциплина: Применение ЭВМ в технологии
лекарственных препаратов
Контрольная работа № 1
Кутепова Оксана АлександровнаШифр: 99311
Курс: 5
Специальность: фармация
г. Днепропетровск
Содержание.
1. Основы квантовой механики атома. Соотношение де Бройля. Уравнение Шредингера..................................................................................................... 3
2. Ионная (гетерополярная) связь. Расчет энергии ионной связи................ 6
3. Теория ковалентной (гомеополярной) связи. Метод валентных связей... 8
4. Теория ковалентной связи. Метод молекулярных орбиталей (МО)...... 12
5. Упрощенный метод МО Хюккеля............................................................ 15
6. Особенности квантово-химических методов............................................ 16
7. Некоторые полуэмпирические методы.................................................... 17
8. Приближения молекулярной механики, лежащие в основе квантово-химических методов.......................................................................................................... 19
Литература.................................................................................................... 21
1. Основы квантовой механики атома. Соотношение де Бройля. Уравнение Шредингера.
Химические процессы сводятся к превращению молекул, т.е. к возникновению и разрушению связей между атомами. Поэтому важнейшей проблемой химии всегда была и остается проблема химического взаимодействия, тесно связанная со строением и свойствами вещества. Современная научная трактовка вопросов химического строения и природы химической связи дается квантовой механикой – теорией движения и взаимодействия микрочастиц (электронов, ядер и т.д.).
Одним из общих свойств материи является ее двойственность. Частицы материи обладают одновременно и корпускулярными и волновыми свойствами. Соотношение "волна – частица" таково, что с уменьшением массы частицы ее волновые свойства все более усиливаются, а корпускулярные – ослабевают. Когда же частица становится соизмеримой с атомом, наблюдаются типичные волновые явления. Одновременно оказывается невозможным описание движения и взаимодействия микрочастиц-волн законами движения тел с большой массой. Первый шаг в направлении создания волновой, или квантовой механики, законы которой объединяют и волновые, и корпускулярные свойства частиц, сделал де Бройлем (1924). Де Бройль высказал гипотезу, что с каждой материальной частицей связан некоторый периодический процесс. Если частица движется, то этот процесс представляется в виде распространяющейся волны, которую называют волной де Дройля, или фазовой волной. Скорость частицы V связана с длиной волны λ соотношением де Бройля:
![]() (1)
(1)
где m – масса частицы (например, электрона);
h – постоянная Планка.
Уравнение (1) относится к свободному движению частиц. Если же частица движется в силовом поле, то связанные с ней волны описываются так называемой волновой функцией. Общий вид этой функции определил Шредингер (1926). Найдем волновую функцию следующим путем. Уравнение, характеризующее напряженность поля Еа плоской монохроматической волны света, можно записать в виде:
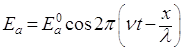 , (2)
, (2)
где Еа0 – амплитуда волны;
ν – частота колебаний;
t – время;
λ – длина волны;
х – координата в направлении распространения волны.
Так как вторые производные от уравнения плоской волны (2), взятые по времени t и координате х, равны соответственно:
![]() , (3)
, (3)
![]() , (4)
, (4)
то ![]()
Подставляя λ=с/ V (с – скорость света), получаем волновое уравнение для плоской световой волны:
![]() , (5)
, (5)
Последующие преобразования основываются на предположениях, что распространение волн де Бройля описывается аналогичным уравнением, и что эти волны становятся стационарными и сферическими. Сначала представим, что по уравнению (5) изменяется значение новой функции ψ от координат (χ, y, z), имеющей смысл амплитуды некоторого колебательного процесса. Тогда, заменяя Еа на ψ, получим волновое уравнение в форме:
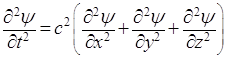 , (6)
, (6)
После исключения t (с помощью (3)) волновое уравнение примет вид:
![]() , (7)
, (7)
где ψ – так называемая волновая функция – величина, периодически изменяющаяся по закону гармонического движения;
ν2 – оператор Лапласа, означающий, что над функцией производится следующее действие:
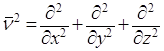 .
.
Будем считать, что волновое уравнение (7) описывает движение частицы. Тогда λ – длина фазовой волны, а ψ – амплитуда фазовой волны в любой произвольно взятой точке χ, y, z, характеризующей местоположение частицы. Длину и амплитуду фазовой волны можно связать с массой и энергией частицы. Если частица движется в потенциальном поле, то ее полная энергия Е складывается из кинетической энергии Ек = mV2/2 и потенциальной энергии Еп. Отсюда
½mV2 – Е – Еп или m2V2 = 2m(E – Eп).
Учитывая соотношение де Бройля, запишем
m2V2 = h2/λ2 и λ2 = h2/2m (E – Eп)
и представим волновое уравнение в следующем виде:
![]() (8)
(8)
В этой форме волновое уравнение называется уравнением Шредингера. Оно является основным уравнением квантовой механики.
Уравнение Шредингера – дифференциальное уравнение в частных производных и может иметь множество решений. Однако физический смысл имеют лишь те ψ-функции (так называемые собственные функции), которые удовлетворяют ряду условий. Во-первых, эти функции должны быть непрерывными, конечными, однозначными и обращаться в нуль на бесконечном расстоянии. Наложение перечисленных условий называется нормированием ψ-функции. Во-вторых, собственным ψ-функциям соответствуют не любые, а только дискретные значения полной энергии Е. Как дискретные значения энергии, так и вид собственных ψ-функций определяется совокупностью квантовых чисел n, l, m, которые хотя и не содержатся в самом уравнении Шредингера, но вводятся в него при решении. Таким образом, квантование энергии естественно и неизбежно вытекает из основных свойств материальных объектов и не нуждается в особом постулировании, которое было сделано Н. Бором при разработке планетарной модели атома.
2. Ионная (гетерополярная) связь. Расчет энергии ионной связи.В зависимости от свойств элементов образующие химическую связь электроны могут находиться в различных энергетических и пространственных состояниях, в результате чего в молекулах возникают разные типы связей. С целью классификации выделяют обычно два основных типа связи – ионную и ковалентную. Однако это разделение условно и не отражает многообразия форм химического движения.
Связь называется ионной в том случае, когда между двумя атомами или группами атомов сильно преобладает электростатическое взаимодействие.
Сродством атома к электрону называется количество энергии Е, которое выделяется при присоединении электрона к нейтральному атому или отрицательному иону
![]()
Полусумма энергии ионизации J и энергии сродства к электрону Е, называется электроотрицательностью χ атома, т.е. χ= ½ (J+E).
Энергия ионизации и сродство к электрону могут быть вычислены квантово-механическим путем для конкретных оболочек атомов, т.е. с учетом степени гибридизации связей и заселенности орбиталей. В связи с этим все шире используется понятие орбитальной электроноотрицательности (ОЭО), с помощью которого оценивается способность атома в молекуле к притяжению электрона на данную орбиталь. Метод ЭО позволяет рассчитать эффективные заряды, которые определяются только нормальными валентными связями атомов. В случае дополнительных эффектов (водородные связи, трансвлияние, дативное взаимодействие и т.п.) вычисленные значения зарядов атомов могут существенно отличаться от экспериментальных.
Энергию образования U гетерополярного соединения из атомов можно найти теоретически. Энергия молекулы как функция расстояния r между одновалентными ионами выражается уравнением:
![]()
В этом уравнении разность энергии ионизации первого атома J1 и энергии сродства к электрону второго атома Е2 выражает энергию образования ионов. Энергия электростатического притяжения ионов представлена отрицательным значением члена е2/r, а энергия отталкивания – функцией b2/r (обусловлена взаимодействием заполненных электронных оболочек). Постоянная n определяется сжимаемостью кристаллического вещества и обычно равна 10. Значение b можно рассчитать из равновесного значения энергии (минимум энергии, когда r = r0):
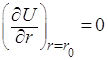 и, следовательно,
и, следовательно,
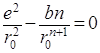 , откуда
, откуда ![]()
Используя это значение b, получим энергию молекулы в равновесном состоянии:
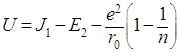 (9)
(9)
Величины, входящие в это уравнение, определяются с помощью спектральных и рентгенографических измерений.
3. Теория ковалентной (гомеополярной) связи. Метод валентных связей.Связь называется ковалентной (гомеополярной), если образующие ее атомы обладают близким сродством к электрону. В этом случае не происходит преимущественной передачи электрона какому-либо атому. Обычно ковалентная связь образуется за счет обобществления электронов, ранее принадлежавших двум отдельным атомам.
Природа ковалентной связи значительно сложнее, чем ионной, и объясняется лишь на основе квантовой механики, причем строго количественное исследование возможно пока что для простейших молекул (Н2, Н2+ и некоторых других). Для сложных соединений решение уравнения Шредингера производится с помощью приближенных методов, дающих чаще всего только качественные результаты.
К наиболее распространенным методам квантовой химии относятся метод валентных связей (электронных пар) и метод молекулярных орбиталей (МО). Конечная цель обоих методов – нахождение энергии и получение из одноэлектронных атомных волновых функций приближенных волновых функций молекул. Значения Е и ψ должны быть такими, чтобы после подстановки уравнение Шредингера превращалось в тождество. Эти методы в ходе математических расчетов широко отражаются на данные физико-химических исследований свойств молекул.
Метод валентных связей (ВС) разработан Гейтлером и Лондоном (1927) при изучении строения молекулы водорода. Метод основан на предположении, что химическая связь образуется парой электронов в процессе сближения и взаимодействия атомов. Молекулу водорода можно изобразить тремя валентными структурами На – Нb, На- – Нb+ и На+ – Нb- с различным расположением (смещением) электронной пары. Взаимодействие ядер а и b и электронов 1 и 2 схематически изображено на рис 1. Так как волновая функция ψ зависит от координат двух электронов, то уравнение Шредингера для такой молекулярной системы принимает вид:
![]() , (10)
, (10)
где ![]() и
и ![]() - операторы Лапласа по координатам (χ1, y1, z1,) первого электрона и координатам (χ2, y2, z2,) второго электрона.
- операторы Лапласа по координатам (χ1, y1, z1,) первого электрона и координатам (χ2, y2, z2,) второго электрона.
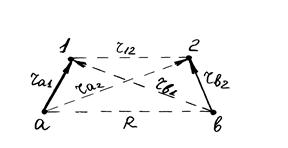
Рис. 1 Схема взаимодействий ядер и электронов в молекуле водорода.
С учетом всевозможных взаимодействий микрочастиц в молекуле Н2 потенциальная энергия находится из равенства:
![]() , (11)
, (11)
где первые два члена в скобках выражают соответственно энергии взаимного отталкивания ядер и электронов, остальные – энергии притяжения электронов к ядрам.
Точное нахождение волновой ψ-функции и минимума полной энергии с помощью уравнений (10) и (11) невозможно даже для такой простой двухэлектронной системы, как молекула Н2, поэтому используется приближенный метод. Сначала оценивают волновую функцию и энергию изолированных атомов, а далее переходят к системе из связанных атомов.
Обозначим волновые функции двух изолированных атомов φа(1) и φb(2). Тогда волновая функция ψІ системы из двух несвязанных атомов выражается произведением ψІ = φа(1) ∙ φb(2).
Допустим, что атомы сблизились на расстояние, достаточное для образования химической связи, и что при этом функция ψІ не изменилась и близка к истинной. Однако в новом состоянии принадлежность каждого электрона к любому из ядер равновероятна, и можно записать, что
ψІІ = φа(2) ∙ φb(1). Тога ψ± - функция молекулы Н2 является линейной комбинацией двух атомных функций:
ψ± = с1ψІ + с2ψІІ, (12)
где с1 и с2 – некоторые постоянные.
Уравнение (12) является общим решением уравнения (10). Конкретное его решение состоит в нахождении значений с1 и с2 и далее по ψ± приближенного значения энергии Е±. Искомую ψ-функцию выбирают с помощью вариационного метода, которые дает
с1 = ± с2 и ψ± = ψІ + ψІІ.
Функция ψ+ = φа(1)∙φb(2) + φа(2)∙φb(1), не изменяющая знак при перестановке электронов, называется симметричной. Меняющая знак функция ψ- = φа(1)∙φb(2) – φа(2)∙φb(1), называется асимметричной.
Выражение для энергии, которая отвечает функции ψ±, имеет вид:
![]() , (13)
, (13)
Уравнение (13) показывает, как должно изменяться значение полной энергии для симметричной и асимметричной функции. Величины J, K и S обозначают три интеграла:
1) кулоновский интеграл J выражает энергию взаимодействия зарядов при отсутствии обмена электронов между ядрами:
![]() ,
,
где ![]() можно рассматривать как члены гамильтониана, выражающие взаимодействие между атомами;
можно рассматривать как члены гамильтониана, выражающие взаимодействие между атомами;
2) обменный интеграл К характеризует уменьшение энергии системы, связанное с обменом электронов местами:
![]() ;
;
3) интеграл перекрывания S отвечает перекрыванию волновых функций соединяющихся атомов:
![]()
Сравнением величин интегралов можно показать, что на расстоянии
R = r0 |К|>>|J| (при этом обе величины отрицательны). S ≈ 0,6, а решение уравнения (13) дает два значения энергии Е+ < 2Е0 и Е- > 2Е0. Следовательно, образование химической связи (минимум энергии Е+) характеризуется функцией ψ+.
Вид волновой ψ-функции каждого электрона зависит только от трех квантовых чисел n, l, m. Очевидно, электроны в молекуле, состояние которых описывается симметричной ψ+-функцией, должны иметь различные спиновые квантовые числа – их спины противоположно направлены, или антипараллельны. Наоборот, ψ--функция отвечает состоянию электронов с одинаково направленными или параллельными спинами.
Таким образом, метод ВС приводит к выводу, что основное значение при образовании химической связи имеет обменное взаимодействие зарядов, удовлетворяющее условию антипараллельности спинов электронов.
Метод ВС позволяет решать ряд задач, связанных с изучением молекул. С его помощью получены ценные сведения о строении и свойствах бензола и его соединений, некоторых многоатомных молекул ионов. Используемые в методе валентные схемы наглядны и близки к классическим химическим формулам. Однако, составляющий основу метода принцип обязательного спаривания электронов с антипараллельными спинами справедлив лишь для S-электронов. С точки зрения метода ВС невозможно понять свойства парамагнитных молекул (например, О2), свойства многих сопряженных и ароматических систем, а также большинства неорганических молекул. Встречаются немалые трудности и при решении других задач.
4. Теория ковалентной связи. Метод молекулярных орбиталей (МО).Метод МО начал разрабатываться в 30-х годах ХХ века в работах ряда ученых (Гунд, Малмекен и др.). В этом методе каждый электрон рассматривается движущимся в поле всех электронов и всех ядер молекулы. Как и в других методах квантовой химии при этом используется одноэлектронное приближение, согласно которому каждый электрон описывается отдельной волновой функцией, а из них составляется полная волновая функция молекулы. Одновременно учитывается адиабатическое приближение Борна-Оппенгеймера; движение электронов молекулы ввиду относительной замедленности колебательных движений массивных ядер рассматривается в поле фиксированных ядер.
Для построение молекулярных орбиталей используется вариант метода, называемый линейной комбинацией атомных ордиталей – молекулярные орбитали (ЛКАО – МО). В его основе лежит способ получения одноэлектронных молекулярных орбиталей (МО) в виде линейной комбинации атомных орбиталей (ЛКАО). Если по-прежнему для двухатомной молекулы (например, Н2) обозначить волновые функции атомов φа и φb (атомные орбитали), то в общем виде их линейные комбинации будут описывать движение каждого электрона в молекуле следующим образом:
![]() ,
,
где і – номер МО;
j – номер АО;
сj – изменяемые параметры, учитывающие долю каждой из суммируемых орбиталей (находятся из условия минимума энергии).
Далее в методе МО допускается, что волновая функция, описывающая состояние многоатомной молекулы, может быть представлена как произведение волновых функций одноэлектронных МО:
ψI = ψ1ψ2 = [φa(1) + φb(1)] [φа(2) ∙ φb(2)].
Таким образом, состояние молекулы Н2 в методе МО определяется совокупностью ряда слагаемых:
ψI = φa(1)∙φb(2) + φа(2)∙φb(1) + φа(1)∙φа(2) + φb(1)∙φb(2)
Сравнение последнего соотношения с применявшимися в методе ВС функциями ψ± показывает, что в методе МО волновая функция ψI отличается третьим и четвертым членом. Оба эти члена характеризуют случаи, когда два электрона находятся либо у одного, либо у другого ядра, т.е. соответствуют ионным состояниям молекулы На- – Нb+ и На+ – Нb-. В действительности роль таких состояний в характеристике МО невелика и составляет около 6 % от энергии обменного взаимодействия, которым определяется ковалентная связь.
Последующие расчеты в методе МО, как и в методе ВС, направлены на определение энергии системы. Здесь так же, как и в методе ВС, получают два значения (уровня) энергии. Первый уровень отвечает соединению атомов и образованию химической связи, поэтому первая орбиталь называется связывающей. Второй уровень характеризует отталкивание, и соответствующая орбиталь называется разрыхляющей. Схема образования химической связи в молекуле Н2 в методе МО изображена на рис. 2.
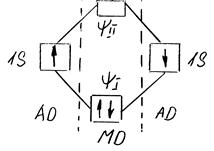
Рис. 2. Схема образования ковалентной связи в Н2.
На рисунке показано, что образование химической ковалентной связи происходит за счет спаривания двух электронов с противоположной ориентацией спинов. Ячейка, соответствующая состоянию этих электронов, расположена ниже ячеек АО, так как энергия МО ψІ меньше, чем у исходных АО. В то же время энергия МО ψІІ больше и ячейка свободна от электронов. При возбуждении молекулы и появлении на верхней МО так называемых разрыхляющих электронов система распадается на отдельные атомы.
Для составления схем образования химических связей более сложных молекул необходимо руководствоваться рядом положений. Во-первых, принцип построения АО повторяет картину заполнения электронных оболочек в атомах. В соответствии с принципом Паули и правилом Гунда заполнение электронами начинается с низшей АО.
В образовании химической связи участвуют лишь валентные электроны. Во-первых, число МО в сложной молекуле увеличивается пропорционально числу атомов в молекуле. В общем случае при использовании NАО образуется NМО. Из них всегда N/2 является связывающими МО и N/2 – разрыхляющими МО. Кратность связи устанавливается избытком числа связывающих электронов (т.е. разностью между количеством связывающих и разрыхляющих электронов), поделенной на 2.
Рассмотрим более сложный случай образования МО на примере молекулы кислорода. Здесь два разрыхляющих электрона расположены на вырожденном уровне π*2р. В соответствии с правилом Гунда они неспарены и имеют параллельные спины. Подсчет кратности связи дает валентность 2, однако из рисунка видно, что обычно принимаемая валентная схема О = О неверна.
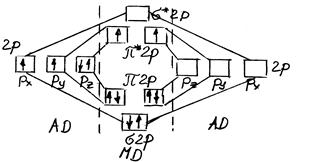
Рис. 3 Схема образования МО в молекуле О2.
В действительность в молекуле О2 в основном ее состоянии двойная связь образуется из трехкратной за счет ее разрыхления двумя электронами. Отсюда видно, что молекула О2 имеет два свободных электрона. Следовательно, кислород должен обладать парамагнитными свойствами. Этот вывод вполне согласуется с опытом.
Метод МО широко используется для изучения химических свойств и реакционной способности простых и сложных соединений. Он, в частности, применяется при исследовании молекул с сопряженными связями, а также при описании свойств многих неорганических соединений.
5. Упрощенный метод МО Хюккеля.Вариант метода МО, предложенный Хюккелем (МОХ), содержит довольно грубые допущения и, как правило, не позволяет осуществлять точные расчеты. Несмотря на это, он часто используется в органической химии при качественном рассмотрении строения соединений с сопряженными связями, для сопоставления их свойств и предсказывания реакционной способности.
Главной особенностью метода МОХ является π-электронное приближение, в соответствии с которым молекулы, имеющие σ- и π-связи, рассчитывают лишь с учетом π-электронов. σ-электроны предполагаются локализованными возле приближенных ядер и не рассматриваются. Для всех атомов, образующих π-связи, π-электроны считаются общими и делокализованными во всем пространстве, занимаемом этими атомами. Волновая функция и уравнение Шредингера записываются лишь для π-электронов, а σ-электроны включаются в ядерный остов, движение которого не учитывается.
Кроме π-электронного приближения, в методе МОХ используется следующими допущениями:
1) кулоновские интегралы одинаковых атомов считают равными:
Jii = Jij = α;
2) резонансные интегралы одинаковых соседних атомов считают равными друг другу, а более удаленных – равными нулю:
kij = β (при j ± 1); kij = 0 (при i>j+1 и i<j+1).
3) интегралы непрерывания принимают равными нулю Sij = 0 (при i ≠ j);
нормировочные интегралы Sii = Sij = 1.
6. Особенности квантово-химических методов.Методы современной квантовой химии распространяются на все более сложные объекты.
Общие принципы квантово-химических расчетов во всех случаях остаются сходными. Каждый объект с позиций метода МО считается единой системой, подчиняющейся законам квантовой механики. Обычно применяются адиабатическое и одноэлектронное приближения, вариант ЛКАОМО, вариационный метод с уравнениями Гутана. Кроме метода ССП (самосогласованного поля) и теории возмущений используется целый ряд упрощенных так называемых полуэмпирических методов.
Появление последних связано с тем, что последовательное применение метода МО к различным молекулярным объектам связано с большими вычислительными трудностями. С ростом количества частиц системы сильно увеличивается число членов уравнения Шредингера, отражающих потенциальную энергию их взаимодействия, а потому и количество подлежащих решению волновых уравнений.
В настоящее время наметилось два пути развития квантовой химии. Один из них – неэмпирический – предполагает минимальное привлечение экспериментальных данных и наиболее полный расчет с использованием орбиталей всех электронов исследуемой системы. Его недостатком являются нарастающие вычислительные трудности при увеличении сложности системы.
Другой путь реализуется с помощью различных полуэмпирических методов, которые используют дополнительные приближения – учитывают не все, а лишь валентные электроны или даже часть из них, как в методе МОХ; интегралы, появляющиеся в расчетах, либо принимаются за нуль, либо считаются независящими от положения атомов в молекуле и определяются из опыта или расчетов и т.д. Такие методы не столь сложны и целесообразны для сравнительной оценки свойств однотипных соединений.
7. Некоторые полуэмпирические методы.Из полуэмпирических методов заслуживают внимания метод "объединенного атома" и "метод атомов в молекулах" Эти методы основаны на рассмотрении непрерывной зависимости электронной энергии молекулы от расстояния между ядрами. Если все межъядерные расстояния в молекуле мысленно устремить к нулю, то электронная оболочка молекулы переходит в электронную оболочку т.н. объединенного атома, заряд ядра которого равен сумме зарядов ядер атомов, составляющих молекулу. В методе объединенного атома волновая функция молекулы разлагается в ряд по взаимно ортогональным волновым функциям различных состояний объединенного атома, ядро которого мысленно помещается в центр тяжести положительных зарядов ядер в молекуле. В расчете энергии молекулы при определении значений ряда интегралов используются спектроскопические данные об энергии термов объединенного атома. В методе "атомов в молекулах" электронная волновая функция молекулы разлагается в ряд по волновым функциям, описывающим различное состояние продуктов диссоциации молекулы (атомов или ионов), а в расчете в энергии молекулы используются опытные значения энергии этих продуктов. Привлечение экспериментальных данных атомной спектроскопии позволяет в методе "объединенного атома" и в методе "атомов в молекулах" в значительной мере уменьшить ошибки, связанные с неточностями в учете взаимной зависимости в движении различных электронов (т.н. эффектов электронной корреляции). Однако расчеты по этим методам могут привести к другим, трудно контролируемым погрешностям, что является серьезным ограничением их применимости.
Заслуживают внимания также модельные методы квантовой химии, в которых для описания электронной структуры сложных молекул используются простые модели, отражающие важнейшие особенности электронной структуры реальных объектов. Типичным примером такого рода является модель свободных электронов для π-электронов в сопряженных и ароматических углеводородах. В простейшем варианте этой модели принимается, что π-электроны свободно движутся вдоль цепочки сопряженных связей. Одномерные волновые функции и уровни энергии электронов легко вычисляются:
![]() ,
, ![]() (n =1, 2, … – номер уровня энергии).
(n =1, 2, … – номер уровня энергии).
В первом – адиабатическом – приближении, предложенном М. Борном и Р. Оппенгеймером в 1927 году, полагают, что движение электронов можно рассматривать как независимое от медленного движения ядер, так как массы ядер значительно (на 3-4 порядка) превышают массу электронов. Решение задачи в этом случае разбивается на два этапа: сначала решают уравнение Шредингера только для электронной части гамильтониана при фиксированном положении ядер. При этом волновая функция должна быть асимметричной по отношению к перестановке электронов, т.е. при перестановке двух электронов с одинаковыми спинами полная волновая функция должна менять знак (принцип Паули). Затем решают задачу о движении (колебании) ядер в поле потенциала, полученного при решении предыдущей задачи, при этом получают значения колебательной энергии молекулы.
Основы квантовой теории многоэлектронных систем были заложены в работах В.Гейзенберга, В. Гайтлера и Ф. Лондона (1926 – 1927 г.г.). Они показали, что существование, устойчивость и свойства этих систем невозможно объяснить в рамках классических представлений. Согласно Гайтлеру и Лондону, связывание между атомами и молекулами в молекуле водорода обусловлено т.н. обменным взаимодействием.
Дальнейшее развитие теории многоэлектронных атомов связано с методом самосогласованного поля, предложенного в 1927 году Д.Р.Хартли. В нем взаимодействие каждого из электронов со всеми остальными заменяется взаимодействием с усредненным полем, создаваемым остальными электронами. В 1930 году В.А.Фок усовершенствовал метод Хартли, использовав для многоэлектронной волновой функции представление в виде слейтеровского детерминанта:
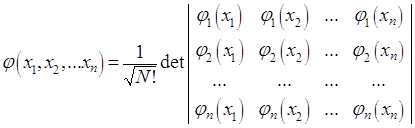 ,
,
где φj(xj) – одноэлектронная спин-орбиталь, xj = (rj, αj), где rj – пространственные координаты, αj – спиновые координаты.
Такой вид волновой функции позволяет учесть принцип Паули. Одноэлектронные функции (орбитали) находят, решая уравнение Хартли-Фока:
Fφi = εiφi,
где F – оператор, называемый фоксианом;
εi – энергии i-той заполненной орбитали.
Система уравнений Хартли-Фока является системой нелинейных интегро-дифференциальных уравнений, которые можно решать методом итераций.
Литература.
1. Минкин В.И. и др. Квантовая химия органических соединений. – М. Химия, 1986.
2. Кларк Т. Молекулярная механика. – М.: "Мир", 1990.
3. Краснов К.С. Молекулы и химическая связь. – М.: Химия, 1984.
4. Шустович С.М. Химическая связь. – М.: Наука, 1973.
Похожие работы

... , основанной на поглощении атомами рентгеновского излучения. Ультрафиолетовая спектрофотометрия — наиболее простой и широко применяемый в фармации абсорбционный метод анализа. Его используют на всех этапах фармацевтического анализа лекарственных препаратов (испытания подлинности, чистоты, количественное определение). Разработано большое число способов качественного и количественного анализа ...
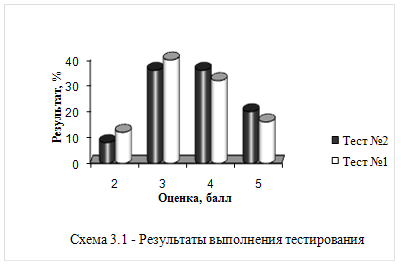
... Br, Cl, F. Результаты тестирования были занесены в таблицы 3.1 и 3.2. На втором этапе педагогического эксперимента школьникам была предложена анкета для изучения их мнения по применению компьютерных технологий на уроках химии. 1. Какие современные средства обучения используются в преподавании химии в вашей школе? a) компьютеры, b) видеозаписи, ...
... на фармакологический эффект, усложняет процесс изыскания новых Л В. Тем не менее современные методы исследования позволили определить предпосылки решения этой важной проблемы. 7 Предпосылки создания новых лекарственных веществ Изыскание новых ЛВ осуществляют различными путями. Ведущим направлением являются исследования в области модификации структуры известных природных БАВ. Одним из ...
... названия. В качестве основного синонима будут также приводиться торговые названия, под которыми JIC зарегистрировано или производится в Российской Федерации. 4 Методологические основы классификации лекарственных средств Количество ЛС в мире непрерывно возрастает. На фармацевтическом рынке в России в настоящее время обращается более I8 ООО наименований ЛС, что в 2,5 раза больше, чем в 1992 г. ...
0 комментариев