Вступление
Глава 1. Основные принципы фармацевтического анализа
1.1 Критерии фармацевтического анализа
1.2 Ошибки, возможные при проведении фармацевтического анализа
1.3 Общие принципы испытаний подлинности лекарственных веществ
1.4 Источники и причины недоброкачественности лекарственных веществ
1.5 Общие требования к испытаниям на чистоту
1.6 Методы фармацевтического анализа и их классификация
Глава 2. Физические методы анализа
2.1 Проверка физических свойств или измерение физических констант лекарственных веществ
2.2 Установление рН среды
2.3 Определение прозрачности и мутности растворов
2.4 Оценка химических констант
Глава 3. Химические методы анализа
3.1 Особенности химических методов анализа
3.2 Гравиметрический (весовой) метод
3.3 Титриметрические (объемные) методы
3.4 Газометрический анализ
3.5 Количественный элементный анализ
Глава 4. Физико-химические методы анализа
4.1 Особенности физико-химических методов анализа
4.2 Оптические методы
4.3 Абсорбционные методы
4.4 Методы, основанные на испускании излучения
4.5 Методы, основанные на использовании магнитного поля
4.6 Электрохимические методы
4.7 Методы разделения
4.8 Термические методы анализа
Глава 5. Биологические методы анализа1
5.1 Биологический контроль качества лекарственных средств
5.2 Микробиологический контроль лекарственных средств
Выводы
Список использованной литературы
Вступление
Фармацевтический анализ — это наука о химической характеристике и измерении биологически активных веществ на всех этапах производства: от контроля сырья до оценки качества полученного лекарственного вещества, изучения его стабильности, установления сроков годности и стандартизации готовой лекарственной формы. Фармацевтический анализ имеет свои специфические особенности, отличающие его от других видов анализа. Эти особенности заключаются в том, что анализу подвергают вещества различной химйческой природы: неорганические, элементорганические, радиоактивные, органические соединения от простых алифатических до сложных природных биологически активных веществ. Чрезвычайно широк диапазон концентраций анализируемых веществ. Объектами фармацевтического анализа являются не только индивидуальные лекарственные вещества, но и смеси, содержащие различное число компонентов. Количество лекарственных средств с каждым годом увеличивается. Это вызывает необходимость разработки новых способов анализа.
Способы фармацевтического анализа нуждаются в систематическом совершенствовании в связи с непрерывным повышением требований к качеству лекарственных средств, причем растут требования как к степени чистоты лекарственных веществ, так и к количественному содержанию. Поэтому необходимо широкое использование не только химических, но и более чувствительных физико-химических методов для оценки качества лекарств.
К фармацевтическому анализу предъявляют высокие требования. Он должен быть достаточно специфичен и чувствителен, точен по отношению к нормативам, обусловленным ГФ XI, ВФС, ФС и другой НТД, выполняться в короткие промежутки времени с использованием минимальных количеств испытуемых лекарственных препаратов и реактивов.
Фармацевтический анализ в зависимости от поставленных задач включает различные формы контроля качества лекарств: фармакопейный анализ, постадийный контроль производства лекарственных средств, анализ лекарственных форм индивидуального изготовления, экспресс-анализ в условиях аптеки и биофармацевтический анализ.
Составной частью фармацевтического анализа является фармакопейный анализ. Он представляет собой совокупность способов исследования лекарственных препаратов и лекарственных форм, изложенных в Государственной фармакопее или другой нормативно-технической документации (ВФС, ФС). На основании результатов, полученных при выполнении фармакопейного анализа, делается заключение о соответствии лекарственного средства требованиям ГФ или другой нормативно-технической документации. При отклонении от этих требований лекарство к применению не допускают.
Заключение о качестве лекарственного средства можно сделать только на основании анализа пробы (выборки). Порядок ее отбора указан либо в частной статье, либо в общей статье ГФ XI (вып. 2). Отбор пробы производят только из неповрежденных укупоренных и упакованных в соответствии с требованиями НТД упаковочных единиц. При этом должны строго соблюдаться требования к мерам предосторожности работы с ядовитыми и наркотическими лекарственными средствами, а также к токсичности, огнеопасности, взрывоопасности, гигроскопичности и другим свойствам лекарств. Для испытания на соответствие требованиям НТД проводят многоступенчатый отбор проб. Число ступеней определяется видом упаковки. На последней ступени (после контроля по внешнему виду) берут пробу в количестве, необходимом для четырех полных физико-химических анализов (если проба отбирается для контролирующих организаций, то на шесть таких анализов).
Из расфасовки "ангро" берут точечные пробы, взятые в равных количествах из верхнего, среднего и нижнего слоев каждой упаковочной единицы. После установления однородности все эти пробы смешивают. Сыпучие и вязкие лекарственные средства отбирают пробоотборником, изготовленным из инертного материала. Жидкие лекарственные средства перед отбором проб тщательно перемешивают. Если это делать затруднительно, то отбирают точечные пробы из разных слоев. Отбор выборок готовых лекарственных средств осуществляют в соответствии с требованиями частных статей или инструкций по контролю, утвержденных МЗ РФ.
Выполнение фармакопейного анализа позволяет установить подлинность лекарственного средства, его чистоту, определить количественное содержание фармакологически активного вещества или ингредиентов, входящих в состав лекарственной формы. Несмотря на то, что каждый из этих этапов имеет свою конкретную цель, их нельзя сматривать изолированно. Они взаимосвязаны и взаимно дополняют друг друга. Так, например, температура плавления, растворимость, рН среды водного раствора и т.д. являются критериями как подлинности, так и чистоты лекарственного вещества.
Глава 1. Основные принципы фармацевтического анализа 1.1 Критерии фармацевтического анализа
На различных этапах фармацевтического анализа в зависимости от поставленных задач имеют значение такие критерии, как избирательность, чувствительность, точность, время, затраченное на выполнение анализа, израсходованное количество анализируемого препарата (лекарственной формы).
Избирательность метода очень важна при проведении анализа смесей веществ, поскольку дает возможность получать истинные значения каждого из компонентов. Только избирательные методики анализа позволяют определять содержание основного компонента в присутствии продуктов разложения и других примесей.
Требования к точности и чувствительности фармацевтического анализа зависят от объекта и цели исследования. При испытании степени чистоты препарата используют методики, отличающиеся высокой чувствительностью, позволяющие устанавливать минимальное содержание примесей.
При выполнении постадийного контроля производства, а также при проведении экспресс-анализа в условиях аптеки важную роль имеет фактор времени, которое затрачивается на выполнение анализа. Для этого выбирают методы, позволяющие провести анализ в наиболее короткие промежутки времени и вместе с тем с достаточной точностью.
При количественном определении лекарственного вещества используют метод, отличающийся избирательностью и высокой точностью. Чувствительностью метода пренебрегают, учитывая возможность выполнения анализа с большой навеской препарата.
Мерой чувствительности реакции является предел обнаружения. Он означает наименьшее содержание, при котором по данной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью. Термин ''предел обнаружения" введен вместо такого понятия, как "открываемый минимум", им пользуются также взамен термина "чувствительность". На чувствительность качественных реакций оказывают влияние такие факторы, как объемы растворов реагирующих компонентов, концентрации реактивов, рН среды, температура, продолжительность опыта. Это следует учитывать при разработке методик качественного фармацевтического анализа. Для установления чувствительности реакций все шире используют показатель поглощения (удельный или молярный), устанавливаемый спектрофотометрическим методом. В химическом анализе чувствительность устанавливают по величине предела обнаружения данной реакции. Высокой чувствительностью отличаются физико-химические методы анализа. Наиболее высокочувствительны радиохимические и масс-спектральный методы, позволяющие определять 10-8—10-9% анализируемого вещества, полярографические и флуориметрические 10-6—10-9%; чувствительность спектрофотометрических методов Ю-3—10-6%, потенциометрических 10-2%.
Термин "точность анализа" включает одновременно два понятия: воспроизводимость и правильность полученных результатов. Воспроизводимость характеризует рассеяние результатов анализа по сравнению со средним значением. Правильность отражает разность между действительным и найденным содержанием вещества. Точность анализа у каждого метода различна и зависит от многих факторов: калибровки измерительных приборов, точности отвешивания или отмеривания, опытности аналитика и т.д. Точность результата анализа не может быть выше, чем точность наименее точного измерения.
Так, при вычислении результатов титриметрических определений наименее точная цифра — количество миллилитров титранта, израсходованного на титрование. В современных бюретках в зависимости от класса их точности максимальная ошибка отмеривания около ±0,02 мл. Ошибка от натекания тоже равна ±0,02 мл. Если при указанной общей ошибке отмеривания и натекания ±0,04 мл на титрование расходуется 20 мл титранта, то относительная ошибка составит 0,2%. При уменьшении навески и количества миллилитров титранта точность соответственно уменьшается. Таким образом, титриметрическое определение можно выполнять с относительной погрешностью ±(0,2—0,3)%.
Точность титриметрических определений можно повысить, если пользоваться микробюретками, применение которых значительно уменьшает ошибки от неточного отмеривания, натекания и влияния температуры. Погрешность допускается также при взятии навески.
Отвешивание навески при выполнении анализа лекарственного вещества осуществляют с точностью до ±0,2 мг. При взятии обычной для фармакопейного анализа навески 0,5 г препарата и точности взвешивания ±0,2 мг относительная ошибка будет равна 0,4%. При анализе лекарственных форм, выполнении экспресс-анализа такая точность при отвешивании не требуется, поэтому навеску берут с точностью ±(0,001—0,01) г, т.е. с предельной относительной ошибкой 0,1—1%. Это можно отнести и к точности отвешивания навески для колориметрического анализа, точность результатов которого ±5%.
1.2 Ошибки, возможные при проведении фармацевтического анализаПри выполнении количественного определения любым химическим или физико-химическим методом могут быть допущены три группы ошибок: грубые (промахи), систематические (определенные) и случайные (неопределенные).
Грубые ошибки являются результатом просчета наблюдателя при выполнении какой-либо из операций определения или неправильно выполненных расчетов. Результаты с грубыми ошибками отбрасываются как недоброкачественные.
Систематические ошибки отражают правильность результатов анализа. Они искажают результаты измерений обычно в одну сторону (положительную или отрицательную) на некоторое постоянное значение. Причиной систематических ошибок в анализе могут быть, например, гигроскопичность препарата при отвешивании его навески; несовершенство измерительных и физико-химических приборов; опытность аналитика и т.д. Систематические ошибки можно частично устранить внесением поправок, калибровкой прибора и т.д. Однако всегда необходимо добиваться того, чтобы систематическая ошибка была соизмерима с ошибкой прибора и не превышала случайной ошибки.
Случайные ошибки отражают воспроизводимость результатов анализа. Они вызываются неконтролируемыми переменными. Среднее арифметическое случайных ошибок стремится к нулю при постановке большого числа опытов в одних и тех же условиях. Поэтому для расчетов необходимо использовать не результаты единичных измерений, а среднее из нескольких параллельных определений.
Правильность результатов определений выражают абсолютной ошибкой и относительной ошибкой.
Абсолютная ошибка представляет собой разность между полученным результатом и истинным значением. Эта ошибка выражается в тех же единицах, что и определяемая величина (граммах, миллилитрах, процентах).
Относительная ошибка определения равна отношению абсолютной ошибки к истинному значению определяемой величины. Выражают относительную ошибку обычно в процентах (умножая полученную величину на 100). Относительные ошибки определений физико-химическими методами включают как точность выполнения подготовительных операций (взвешивание, отмеривание, растворение), так и точность выполнения измерений на приборе (инструментальная ошибка).
Значения относительных ошибок находятся в зависимости от того, каким методом выполняют анализ и что представляет собой анализируемый объект — индивидуальное вещество или многокомпонентную смесь. Индивидуальные вещества можно определять при анализе спек- трофотометрическим методом в УФ- и видимой областях с относительной погрешностью ±(2—3)%, ИК-спектрофотометрией ±(5—12)%, газо- жидкостцой хроматографией ±(3—3,5)%; полярографией ±(2—3)%; потенциометрией ±(0,3—1)%.
При анализе многокомпонентных смесей относительная погрешность определения этими методами возрастает примерно в два раза. Сочетание хроматографии с другими методами, в частности использование хроматооптических и хроматоэлектрохимических методов, позволяет выполнять анализ многокомпонентных смесей с относительной погрешностью ±(3—7)%.
Точность биологических методов намного ниже, чем химических и физико-химических. Относительная ошибка биологических определений достигает 20—30 и даже 50%. Для повышения точности в ГФ XI введен статистический анализ результатов биологических испытаний.
Относительная ошибка определения может быть уменьшена за счет увеличения числа параллельных измерений. Однако эти возможности имеют определенный предел. Уменьшать случайную ошибку измерений, увеличивая число опытов, целесообразно до тех пор, пока она станет меньше систематической. Обычно в фармацевтическом анализе выполняют 3—6 параллельных измерений. При статистической обработке результатов определений с целью получения достоверных результатов выполняют не менее семи параллельных измерений.
1.3 Общие принципы испытаний подлинности лекарственных веществИспытание на подлинность — это подтверждение идентичности анализируемого лекарственного вещества (лекарственной формы), осуществляемое на основе требований Фармакопеи или другой нормативно-технической документации (НТД). Испытания выполняют физическими, химическими и физико-химическими методами. Непременным условием объективного испытания подлинности лекарственного вещества является идентификация тех ионов и функциональных групп, входящих в структуру молекул, которые обусловливают фармакологическую активность. С помощью физических и химических констант (удельного вращения, рН среды, показателя преломления, УФ- и ИК-спектра) подтверждают и другие свойства молекул, оказывающие влияние на фармакологический эффект. Применяемые в фармацевтическом анализе химические реакции сопровождаются образованием окрашенных соединений, выделением газообразных или нерастворимых в воде соединений. Последние можно идентифицировать по температуре плавления.
1.4 Источники и причины недоброкачественности лекарственных веществОсновные источники технологических и специфических примесей — аппаратура, исходное сырье, растворители и другие вещества, которые используют при получении лекарственных средств. Материал, из которого изготовлена аппаратура (металл, стекло), может служить источником примесей тяжелых металлов и мышьяка. При плохой очистке в препаратах могут содержаться примеси растворителей, волокна тканей или фильтровальной бумаги, песок, асбест и т.д., а также остатки кислот или щелочей.
На качество синтезируемых лекарственных веществ могут оказывать влияние различные факторы.
Технологические факторы — первая группа факторов, оказывающих влияние в процессе синтеза лекарственного вещества. Степень чистоты исходных веществ, температурный режим, давление, рН среды, растворители, применяемые в процессе синтеза и для очистки, режим и температура сушки, колеблющаяся даже в небольших пределах, — все эти факторы могут привести к появлению примесей, которые накапливаются от одной к другой стадии. При этом могут происходить образование продуктов побочных реакций или продуктов распада, процессы взаимодействия исходных и промежуточных продуктов синтеза с образованием таких веществ, от которых трудно затем отделить конечный продукт. В процессе синтеза возможно также образование различных таутомерных форм как в растворах, так и в кристаллическом состоянии. Так, например, многие органические соединения могут существовать в амидной, имидной и других таутомерных формах. Причем нередко в зависимости от условий получения, очистки и хранения лекарственное вещество может представлять собой смесь двух таутомеров или других изомеров, в том числе оптических, различающихся по фармакологической активности.
Вторая группа факторов — образование различных кристаллических модификаций, или полиморфизм. Около 65% лекарственных веществ, относящихся к числу барбитуратов, стероидов, антибиотиков, алкалоидов и др., образуют по 1—5 и более различных модификаций. Остальные дают при кристаллизации стабильные полиморфные и псевдополиморфные модификации. Они различаются не только по физико-химическим свойствам (температуре плавления, плотности, растворимости) и фармакологическому действию, но имеют различную величину свободной поверхностной энергии, а следовательно, неодинаковую устойчивость к действию кислорода воздуха, света, влаги. Это вызвано изменениями энергетических уровней молекул, что оказывает влияние на спектральные, термические свойства, растворимость и абсорбцию лекарственных веществ. Образование полиморфных модификаций зависит от условий кристаллизации, используемого при этом растворителя, температуры. Превращение одной полиморфной формы в другую происходит при хранении, сушке, измельчении.
В лекарственных веществах, получаемых из растительного и животного сырья, основными примесями являются сопутствующие природные соединения (алкалоиды, ферменты, белки, гормоны и др.). Многие из них очень сходны по химическому строению и физико-химическим свойствам с основным продуктом экстракции. Поэтому очистка его представляет большую сложность.
Большое влияние на загрязнение примесями одних лекарственных препаратов другими может оказать запыленность производственных помещений химико-фармацевтических предприятий. В рабочей зоне этих помещений при условии получения одного или нескольких препаратов (лекарственных форм) все они могут содержаться в виде аэрозолей в воздухе. При этом происходит так называемое "перекрестное загрязнение".
Всемирной организацией здравоохранения (ВОЗ) в 1976 г. были разработаны специальные правила организации производства и контроля качества лекарственных средств, которые предусматривают условия предотвращения "перекрестного загрязнения".
Важное значение для качества лекарств имеют не только технологический процесс, но и условия хранения. На доброкачественность препаратов оказывает влияние излишняя влажность, которая может привести к гидролизу. В результате гидролиза образуются основные соли, продукты омыления и другие вещества с иным характером фармакологического действия. При хранении препаратов-кристаллогидратов (натрия арсенат, меди сульфат и др.) необходимо, наоборот, соблюдать условия, исключающие потерю кристаллизационной воды.
При хранении и транспортировке препаратов необходимо учитывать воздействие света и кислорода воздуха. Под влиянием этих факторов может происходить разложение, например, таких веществ, как хлорная известь, серебра нитрат, иодиды, бромиды и т.д. Большое значение имеет качество тары, используемой для хранения лекарственных препаратов, а также материал, из которого она изготовлена. Последний тоже может быть источником примесей.
Таким образом, примеси, содержащиеся в лекарственных веществах, можно разделить на две группы: примеси технологические, т.е. внесенные исходным сырьем или образовавшиеся в процессе производства, и примеси, приобретенные в процессе хранения или транспортировки, под воздействием различных факторов (теплоты, света, кислорода воздуха и т.д.).
Содержание тех и других примесей должно строго контролироваться, чтобы исключить присутствие токсичных соединений или наличие индифферентных веществ в лекарственных средствах в таких количествах, которые мешают их использованию для конкретных целей. Иными словами, лекарственное вещество должно иметь достаточную степень чистоты, а следовательно, отвечать требованиям определенной спецификации.
Лекарственное вещество является чистым, если дальнейшая очистка не меняет его фармакологической активности, химической стабильности, физических свойств и биологической доступности.
В последние годы в связи с ухудшением экологической обстановки на наличие примесей тяжелых металлов испытывают и лекарственное растительное сырье. Важность проведения таких испытаний вызвана тем, что при проведении исследований 60 различных образцов растительного сырья установлено содержание в них 14 металлов, в том числе таких токсичных, как свинец, кадмий, никель, олово, сурьма и даже таллий. Их содержание в большинстве случаев значительно превышает установленные ПДК для овощей и фруктов.
Фармакопейный тест на определение примесей тяжелых металлов — один из широко применяемых во всех национальных фармакопеях мира, которые рекомендуют его для исследования не только индивидуальных лекарственных веществ, но и масел, экстрактов, ряда инъекционных лекарственных форм. По мнению Комитета экспертов ВОЗ, такие испытания следует проводить в отношении лекарственных средств, имеющих разовые дозы не менее 0,5 г.
1.5 Общие требования к испытаниям на чистотуОценка степени чистоты лекарственного препарата — один из важных этапов фармацевтического анализа. Все лекарственные препараты независимо от способа получения испытывают на чистоту. При этом устанавливают содержание примесей. Их можно разделить на две группы: примеси, оказывающие влияние на фармакологическое действие лекарственного препарата, и примеси, указывающие на степень очистки вещества. Последние не влияют на фармакологический эффект,, но присутствие их в больших количествах снижает концентрацию и соответственно уменьшает активность препарата. Поэтому фармакопеи устанавливают определенные пределы этих примесей в лекарственных препаратах.
Таким образом, основной критерий доброкачественности лекарственного препарата — наличие допустимых пределов физиологически неактивных примесей и отсутствие токсичных примесей. Понятие отсутствие условно и связано с чувствительностью способа испытания.
Общие требования, которые предъявляются к испытаниям на чистоту, — чувствительность, специфичность и воспроизводимость используемой реакции, а также пригодность ее применения для установления допустимых пределов содержания примесей.
Для испытаний чистоты избирают реакции с такой чувствительностью, которая позволяет определить допустимые пределы примесей в данном лекарственном препарате. Эти пределы устанавливают предварительной биологической проверкой с учетом возможного токсического воздействия примеси.
Определить максимальное содержание примесей в испытуемом препарате можно двумя путями (эталонным и безэталонным). Один из них основан на сравнении с эталонным раствором (стандартом). При этом в одинаковых условиях наблюдают окраску или помутнение, возникающие под действием какого-либо реактива. Второй путь — установление предела содержания примесей по отсутствию положительной реакции. При этом используют химические реакции, чувствительность которых ниже, чем предел обнаружения допустимых примесей.
Для ускорения выполнения испытаний на чистоту, их унификации и достижения одинаковой точности анализа в отечественных фармако- пеях использована система эталонов. Эталон представляет собой образец, содержащий определенное количество открываемой примеси. Установление наличия примесей производят колориметрическим или нефелометрическим методом, сравнивания результаты реакций в растворе эталона и в растворе препарата после добавления одинаковых количеств соответствующих реактивов. Достигаемая при этом точность вполне достаточна, чтобы установить, больше или меньше, чем допустимо, содержится примесей в испытуемом препарате.
При выполнении испытаний на чистоту необходимо строго соблюдать общие указания, предусмотренные фармакопеями. Вода и используемые реактивы не должны содержать ионов, наличие которых устанавливают; одинакового диаметра и бесцветными должны быть пробирки; навески должны отвешиваться с точностью до 0,001 г; реактивы следует добавлять одновременно и в одинаковых количествах как к эталонному, так и к испытуемому раствору; образующуюся опалесценцию наблюдают в проходящем свете на темном фоне, а окраску — в отраженном свете на белом фоне. Если устанавливают отсутствие примеси, то к испытуемому раствору прибавляют все реактивы, кроме основного; затем полученный раствор делят на две равные части и к одной из них прибавляют основной реактив. При сравнении не должно быть заметных различий между обеими частями раствора.
Следует иметь в виду, что последовательность и скорость прибавления реактива влияют на результаты испытаний на чистоту. Иногда необходимо также соблюдать интервал времени, в течение которого следует вести наблюдение за результатом реакции.
Источником примесей при производстве готовых лекарственных форм могут служить плохо очищенные наполнители, растворители и другие вспомогательные вещества. Поэтому степень чистоты этих веществ должна подвергаться тщательному контролю перед использованием их в производстве.
1.6 Методы фармацевтического анализа и их классификацияВ фармацевтическом анализе используются разнообразные методы исследования: физические, физико-химические, химические, биологические. Применение физических и физико-химических методов требует соответствующих приборов и инструментов, поэтому данные методы называют также приборными, или инструментальными.
Использование физических методов основано на измерении физических констант, например, прозрачности или степени мутности, цветности, влажности, температуры плавления, затвердевания и кипения и др.
С помощью физико-химических методов измеряют физические константы анализируемой системы, которые изменяются в результате химических реакций. К этой группе методов относятся оптические, электрохимические, хроматографические.
Химические методы анализа основаны на выполнении химических реакций.
Биологический контроль лекарственных веществ осуществляют на животных, отдельных изолированных органах, группах клеток, на определенных штаммах микроорганизмов. Устанавливают силу фармакологического эффекта или токсичность.
Методики, используемые в фармацевтическом анализе, должны быть чувствительными, специфическими, избирательными, быстрыми и пригодными для экспресс-анализа в условиях аптеки.
Глава 2. Физические методы анализа 2.1 Проверка физических свойств или измерение физических констант лекарственных веществ
Подлинность лекарственного вещества подтверждают; агрегатное состояние (твердое вещество, жидкость, газ); окраска, запах; форма кристаллов или вид аморфного вещества; гигроскопичность или степень выветриваемости на воздухе; устойчивость к воздействию света, кислорода воздуха; летучесть, подвижность, воспламеняемость (жидкостей). Окраска лекарственного вещества — одно из характерных свойств, позволяющее осуществить его предварительную идентификацию.
Определение степени белизны порошкообразных лекарственных средств — физический метод, впервые включенный в ГФ XI. Степень белизны (оттенка) твердых лекарственных веществ можно оценить различными инструментальными методами на основе спектральной характеристики света, отраженного от образца. Для этого измеряют коэффициенты отражения при освещении образца белым светом, полученным от специального источника со спектральным распределением или пропущенным через светофильтры с максимумом пропускания 614 нм (красный) или 459 нм (синий). Можно также измерять коэффициент отражения света, пропущенного через зеленый светофильтр (522 нм). Коэффициент отражения — это отношение величины отраженного светового потока к величине падающего светового потока. Он позволяет определить наличие или отсутствие у лекарственных веществ цветового оттенка по степени белизны и степени яркости. Для белых или белых с сероватым оттенком веществ степени белизны теоретически равна 1. Вещества, у которых она 0,95—1,00, а степени яркости < 0,85, имеют сероватый оттенок.
Более точно оценку белизны лекарственных веществ можно осуществить с помощью спектрофотометров отражения, например СФ-18, выпускаемых ЛОМО (Ленинградским оптико-механическим объединением). Интенсивность цветовых или сероватого оттенков устанавливают по абсолютным коэффициентам отражения. Значения степени белизны и степени яркости являются характеристиками качества белых и белых с оттенками лекарственных веществ. Их допустимые пределы регламентируются в частных статьях.
Более объективным является установление различных физических констант: температуры плавления (разложения), температуры затвердевания или кипения, плотности, вязкости. Важный показатель подлинности — растворимость лекарственного препарата в воде, растворах кислот, щелочей, органических растворителях (эфире, хлороформе, ацетоне, бензоле, этиловом и метиловом спирте, маслах и др.).
Константой, характеризующей гомогенность твердых веществ, является температура плавления. Ее используют в фармацевтическом анализе для установления подлинности и чистоты большинства твердых лекарственных веществ. Известно, что это температура, при которой твердое тело находится в равновесии с жидкой фазой при насыщенной фазе пара. Температура плавления является постоянной величиной для индивидуального вещества. Присутствие даже небольшого содержания примесей изменяет (как правило, снижает) температуру плавления вещества, что позволяет судить о степени его чистоты. Подтвердить индивидуальность исследуемого соединения можно пробой смешанного плавления, так как смесь двух веществ, имеющих одинаковые температуры плавления, плавится при той же температуре.
Для установления температуры плавления ГФ XI рекомендует капиллярный метод, позволяющий подтвердить подлинность и ориентировочно степень чистоты лекарственного препарата. Так как в лекарственных препаратах допускается некоторое содержание примесей (нормируемое ФС или ВФС), то температура плавления может быть выражена не всегда четко. Поэтому большинство фармакопей, в том числе и ГФ XI, под температурой плавления подразумевает интервал температур, при котором происходит процесс плавления испытуемого препарата от появления первых капель жидкости до полного перехода вещества в жидкое состояние. Некоторые органические соединения при нагревании разлагаются. Процесс этот происходит при температуре разложения и зависит от ряда факторов, в частности от скорости нагрева.
Приведенные в частных статьях ГФ (ФС, ВФС) интервалы температур плавления указывают на то, что между началом и окончанием плавления лекарственного вещества интервал не должен превышать 2°С. Если он превышает 2°С, то в частной статье должно быть указано, на какую величину. Если переход вещества из твердого в жидкое состояние нечеткий, то вместо интервала температуры плавления устанавливают температуру, при которой происходит только начало или только окончание плавления. Это значение температуры должно укладываться в интервал, приведенный в частной статье ГФ (ФС, ВФС).
Описание прибора и методик определения температуры плавления приведено в ГФ XI, вып.1 (с. 16). В зависимости от физических свойств применяют различные методы. Один из них рекомендуется для твердых веществ, легко превращаемых в порошок, а два других — для веществ, не растирающихся в порошок (жиры, воск, парафин, вазелин и др.). Следует учитывать, что на точность установления температурного интервала, при котором происходит плавление испытуемого вещества, могут влиять условия подготовки образца, скорость подъема и точность измерения температуры, опытность аналитика.
В ГФ XI, вып. 1 (с. 18) уточнены условия определения температуры плавления и рекомендован новый прибор с диапазоном измерений в пределах от 20 до 360°С (ПТП) с электрическим обогревом. Он отличается наличием стеклянного блока-нагревателя, обогрев которого осуществляется навитой константановой проволокой, оптическим приспособлением и щитком управления с номограммой. Капилляры для этого прибора должны иметь длину 20 см. Прибор ПТП обеспечивает более высокую точность определения температуры плавления. Если получаются расхождения при определении температуры плавления (указанной в частной статье), то следует приводить результаты ее определения на каждом из использованных приборов.
Под температурой затвердевания понимают наиболее высокую, остающуюся в течение короткого времени, постоянную температуру, при которой происходит переход вещества из жидкого состояния в твердое. В ГФ XI, вып. 1 (с. 20) описаны устройство прибора и методика определения температуры затвердевания. По сравнению с ГФ X в нее внесено дополнение, касающееся веществ, способных переохлаждаться.
Температура кипения, или, точнее говоря, температурные пределы перегонки, — это интервал между начальной и конечной температурой кипения при нормальном давлении 760 мм рт.ст. (101,3 кПа). Температуру, при которой в приемник перегнались первые 5 капель жидкости, называют начальной температурой кипения, а температуру, при которой перешло в приемник 95% жидкости, — конечной температурой кипения. Указанные пределы температур можно установить макрометодом и микрометодом. Помимо прибора, рекомендованного ГФ XI, вып. 1 (с. 18), для определения температуры плавления (ПТП) может быть использован прибор для определения температурных пределов перегонки (ТПП) жидкостей, изготавливаемый Клин- ским заводом "Лаборприбор" (ГФ XI, вып. 1, с. 23). Этот прибор обеспечивает получение более точных и воспроизводимых результатов.
Следует учитывать, что температура кипения зависит от атмосферного давления. Температуру кипения устанавливают только у сравнительно небольшого числа жидких лекарственных препаратов: циклопропана, хлорэтила, эфира, фторотана, хлороформа, трихлорэтилена, этанола.
При установлении плотности берут массу вещества определенного объема. Плотность устанавливают с помощью пикнометра или ареометра по методикам, описанным в ГФ XI, вып. 1 (с. 24—26), строго соблюдая температурный режим, так как плотность зависит от температуры. Обычно это достигают термостатированием пикнометра при 20°С. Определенные интервалы значений плотности подтверждают подлинность этилового спирта, глицерина, масла вазелинового, вазелина, парафина твердого, галогенопроизводных углеводородов (хлорэтила, фторотана, хлороформа), раствора формальдегида, эфира для наркоза, амилнитрита и др. ГФ XI, вып. 1 (с. 26) рекомендует устанавливать содержание спирта в препаратах спирта этилового 95, 90, 70 и 40%-ного по плотности, а в лекарственных формах либо дистилляцией с последующим установлением плотности, либо по температуре кипения водно-спиртовых растворов (в том числе настоек).
Дистилляцию осуществляют кипячением определенных количеств спиртоводных смесей (настоек) в колбах, герметически соединенных с приемником. Последний представляет собой мерную колбу вместимостью 50 мл. Собирают 48 мл отгона, доводят его температуру до 20°С и добавляют водой до метки. Плотность отгона устанавливают пикнометром.
При определении спирта (в настойках) по температуре кипения используют прибор, описанный в ГФ XI, вып. 1 (с. 27). Показания термометра снимают через 5 мин после начала кипения, когда температура кипения стабилизируется (отклонения не более ±0,1°С). Полученный результат пересчитывают на нормальное атмосферное давление. Концентрацию спирта вычисляют с помощью таблиц, имеющихся в ГФ XI, вып. 1 (с.28).
Вязкость (внутреннее трение) — физическая константа, подтверждающая подлинность жидких лекарственных веществ. Различают динамическую (абсолютную), кинематическую, относительную, удельную, приведенную и характеристическую вязкость. Каждая из них имеет свои единицы измерения.
Для оценки качества жидких препаратов, имеющих вязкую консистенцию, например глицерина, вазелина, масел, обычно определяют относительную вязкость. Она представляет собой отношение вязкости исследуемой жидкости к вязкости воды, принятой за единицу. Для измерения кинематической вязкости используют различные модификации вискозиметров типа Оствальда и Уббелоде. Кинематическую вязкость обычно выражают в м2 * с-1. Зная плотность исследуемой жидкости, можно затем вычислить динамическую вязкость, которую выражают в Па * с. Динамическую вязкость можно также установить с помощью ротационных вискозиметров различных модификаций типа ''Полимер РПЭ-1И или микрореометров серии ВИР. На измерении скорости падения шарика в жидкости основано устройство вискозиметров типа Гепплера. Они позволяют установить динамическую вязкость. Все приборы должны термостатироваться, так как вязкость в значительной степени зависит от температуры испытуемой жидкости.
Растворимость в ГФ XI рассматривают не как физическую константу, а как свойство, которое может служить ориентировочной характеристикой испытуемого препарата. Наряду с температурой плавления растворимость вещества при постоянной температуре и давлении является одним из параметров, по которому устанавливают подлинность и чистоту практически всех лекарственных веществ.
Методика определения растворимости по ГФ XI основана на том, что навеска предварительно растертого (в необходимых случаях) препарата вносится в отмеренный объем растворителя и непрерывно перемешивается в течение 10 мин при (20±2)°С. Растворившимся считают препарат, в растворе которого в проходящем свете не наблюдается частиц вещества. Если для растворения препарата требуется более 10 мин, то его относят к числу медленно растворимых. Их смесь с растворителем нагревают на водяной бане до 30° С и наблюдают полноту растворения после охлаждения до (20±2)°С и энергичного встряхивания в течение 1—2 мин. Более детальные указания об условиях растворения медленно растворимых лекарственных веществ, а также препаратов, образующих мутные растворы, приведены в частных статьях. Показатели растворимости в различных растворителях указываются в частных статьях. В них оговариваются случаи, когда растворимость подтверждает степень чистоты лекарственного вещества.
В ГФ XI, вып. 1 (с. 149) включен метод фазовой растворимости, который дает возможность осуществлять количественную оценку степени чистоты лекарственного вещества путем точных измерений значений растворимости. Этот метод основан на правиле фаз Гиббса, которое устанавливает зависимость между числом фаз и числом компонентов в условиях равновесия. Суть установления фазовой растворимости заключается в последовательном прибавлении увеличивающейся массы препарата к постоянному объему растворителя. Для достижения состояния равновесия смесь подвергают длительному встряхиванию при постоянной температуре, а эатем с помощью диаграмм определяют содержание растворенного лекарственного вещества, т.е. устанавливают, является ли испытуемый препарат индивидуальным веществом или смесью. Метод фазовой растворимости отличается объективностью, не требует для выполнения дорогостоящего оборудования, знания природы и структуры примесей. Это позволяет использовать его для качественного и количественного анализов, а также для изучения стабильности и получения очищенных образцов препаратов (до степени чистоты 99,5%), Важное достоинство метода — возможность отличать оптические изомеры и полиморфные формы лекарственных веществ. Метод применим ко всем видам соединений, которые образуют истинные растворы.
2.2 Установление рН средыВажную информацию о степени чистоты лекарственного препарата дает значение рН его раствора. По этому значению можно судить о наличии примесей кислых или щелочных продуктов.
Принцип обнаружения примесей свободных кислот (неорганических и органических), свободных щелочей, т.е. кислотности и щелочности, заключается в нейтрализации этих веществ в растворе препарата или в водном экстракте. Нейтрализацию выполняют в присутствии индикаторов (фенолфталеин, метиловый красный, тимолфталеин, бромфеноловый синий и др). О кислотности или щелочности судят либо по окраске индикатора, либо по ее изменению, либо устанавливают количество титрованного раствора щелочи или кислоты, затраченное на нейтрализацию.
Реакция среды (рН) является характеристикой химических свойств вещества. Это важный параметр, который следует устанавливать при выполнении технологических и аналитических операций. Степень кислотности или основности растворов необходимо учитывать при выполнении испытаний чистоты лекарственных препаратов и количественного определения. От значений рН растворов зависят сроки хранения лекарственных веществ, а также осрбенности их применения.
Значение рН ориентировочно (до 0,3 ед.) можно определять с помощью индикаторной бумаги или универсального индикатора. Из многочисленных способов установления значения рН среды ГФ XI рекомендует колориметрический и потенциометрический способы.
Колориметрический способ весьма несложен по выполнению. Он основан на свойстве индикаторов изменять свою окраску при определенных интервалах значений рН среды. Для выполнения испытаний используют буферные растворы с постоянной концентрацией водородных ионов, отличающихся друг от друга на величину рН, равную 0,2. К серии таких растворов и к испытуемому раствору прибавляют одинаковое количество (2—3 капли) индикатора. По совпадению окраски с одним из буферных растворов судят о значении рН среды испытуемого раствора.
В ГФ XI, вып. 1 (с. 116) приведены подробные сведения о приготовлении стандартных буферных растворов для различных областей рН: от 1,2 до 11,4. В качестве реактивов для этой цели используют сочетания различных соотношений растворов хлорида калия, гидрофталата калия, однозамещенного фосфата калия, борной кислоты, тетрабората натрия с соляной кислотой или раствором гидроксида натрия. Вода очищенная, используемая для приготовления буферных растворов, должна иметь рН 5,8—7,0 и быть свободной от примеси углекислого газа.
Потенциометрический способ следует отнести к физико-химическим (электрохимическим) методам. Потенциометрическое определение рН основано на измерении электродвижущей силы элемента, составленного из стандартного электрода (с известным значением потенциала) и индикаторого электрода, потенциал которого зависит от рН испытуемого раствора. Для установления рН среды используют потенциометры или рН-метры различных марок. Их настройку осуществляют с помощью буферных растворов. Потенциометрический способ определения рН отличается от колориметрического более высокой точностью. Он имеет меньше ограничений, может быть применен для определения рН в окрашенных растворах, а также в присутствии окислителей и восстановителей.
В ГФ XI, вып. 1 (с. 113) включена таблица, в которой указаны растворы веществ, используемых в качестве стандартных буферных растворов, для проверки рН-метров. Приведенные в таблице данные позволяют установить зависимость рН этих растворов от температуры.
2.3 Определение прозрачности и мутности растворовПрозрачность и степень мутности жидкости по ГФ X (с. 757) и ГФ XI, вып. 1 (с. 198) устанавливают путем сравнения при вертикальном расположении пробирок испытуемой жидкости с тем же растворителем или с эталонами. Жидкость считают прозрачной, если при ее освещении матовой электролампой (мощностью 40 Вт) на черном фоне не наблюдается присутствие нерастворенных частиц, кроме единичных волокон. По ГФ X эталоны представляют собой взвесь, полученную из определенных количеств белой глины. Эталонами для определения степени мутности по ГФ XI служат взвеси в воде из смесей определенных количеств гидразина сульфата и гекса- метилентетрамина. Вначале готовят 1%-ный раствор гидразина сульфата и 10%-ный раствор гексаметилентетрамина. Смешиванием равных объемов этих растворов получают исходный эталон.
В общей статье ГФ XI приведена таблица, в которой указаны количества основного эталона, необходимые для приготовления эталонных растворов I, II, III, IV. Здесь же указана схема просмотра прозрачности и степени мутности жидкостей.
Окраску жидкостей по ГФ XI, вып. 1 (с. 194) устанавливают путем сравнения испытуемых растворов с равным количеством одного из семи эталонов при дневном отраженном свете на матово- белом фоне. Для приготовления эталонов используют четыре основных раствора, полученных смешением в различных соотношениях исходных растворов хлорида кобальта, дихромата калия, сульфата меди (II) и хлорида железа (III). В качестве растворителя для приготовления основных растворов и эталонов используют раствор серной кислоты (0,1 моль/л).
Бесцветными считают жидкости, не отличающиеся по цвету от воды, а растворы — от соответствующего растворителя.
Адсорбционная способность и дисперсность также являются показателями чистоты некоторых лекарственных препаратов.
Очень часто используют для обнаружения примесей органических веществ испытание, основанное на их взаимодействии с концентрированной серной кислотой. Последняя при этом может выступать в роли окислителя или дегидратирующего средства.
В результате таких реакций образуются окрашенные продукты. Интенсивность полученной окраски не должна превышать соответствующего эталона цветности.
Для установления чистоты лекарственных препаратов широко используют определение золы (ГФ XI, вып.2, с.24). Прокаливанием навески препарата в фарфоровом (платиновом) тигле устанавливают общую золу. Затем после добавления разведенной соляной кислоты определяют золу, нерастворимую в соляной кислоте. Кроме того, определяют также сульфатную золу, получаемую после нагревания и прокаливания навески препарата, обработанной концентрированной серной кислотой.
Один из показателей чистоты органических лекарственных препаратов — содержание остатка после прокаливания.
При установлении чистоты некоторых лекарственных препаратов проверяют также наличие восстанавливающих веществ (по обесцвечиванию раствора перманганата калия), красящих веществ (бесцветность водного извлечения). Обнаруживают также водорастворимые соли (в нерастворимых препаратах), вещества, нерастворимые в этаноле, и примеси, нерастворимые в воде (по эталону мутности).
2.4 Оценка химических константДля оценки чистоты масел, жиров, воска, некоторых сложных эфиров используют такие химические константы, как кислотное число, число омыления, эфирное число, йодное число (ГФ XI, вып. 1, с. 191, 192, 193).
Кислотное число — масса гидроксида калия (мг), которая необходима для нейтрализации свободных кислот, содержащихся в 1 г исследуемого вещества.
Число омыления — масса гидроксида калия (мг), которая необходима для нейтрализации свободных кислот и кислот, образующихся при полном гидролизе сложных эфиров, содержащихся в 1 г исследуемого вещества.
Эфирное число — масса гидроксида калия (мг), которая необходима для нейтрализации кислот, образующихся при гидролизе сложных эфиров, содержащихся в 1 г исследуемого вещества (т.е. разность между числом омыления и кислотным числом).
Йодное число — масса иода (г), которая связывает 100 г исследуемого вещества.
В ГФ XI приведены методики установления указанных констант и способы их расчета.
Глава 3. Химические методы анализа 3.1 Особенности химических методов анализа
Эти методы используются для установления подлинности лекарственных веществ, испытаний их на чистоту и количественного определения.
Для целей идентификации используют реакции, которые сопровождаются внешним эффектом, например изменением окраски раствора, выделением газообразных продуктов, выпадением или растворением осадков. Установление подлинности неорганических лекарственных веществ заключается в обнаружении с помощью химических реакций катионов и анионов, входящих в состав молекул. Химические реакции, применяемые для идентификации органических лекарственных веществ, основаны на использовании функционального анализа.
Чистота лекарственных веществ устанавливается помощью чувствительных и специфичных реакций, пригодных для определения допустимых пределов содержания примесей.
Химические методы оказались самыми надежными и эффективными, они дают возможность выполнить анализ быстро и с высокой достоверностью. В случае сомнения в результатах анализа последнее слово остается за химическими методами.
Количественные методы химического анализа подразделяют на гравиметрический, титриметрический, газометрический анализ и количественный элементный анализ.
3.2 Гравиметрический (весовой) метод
Гравиметрический метод основан на взвешивании осажденного вещества в виде малорастворимого соединения или отгонки органических растворителей после извлечения лекарственного вещества. Метод точен, но длителен, так как предусматривает такие операции, как фильтрование, промывание, высушивание (или прокаливание) до постоянной массы.
Из неорганических лекарственных веществ гравиметрическим методом можно определять сульфаты, переводя их в нерастворимые соли бария, и силикаты, предварительно прокаливая до диоксида кремния.
Рекомендуемые ГФ методики гравиметрического анализа препаратов солей хинина основаны на осаждении основания этого алкалоида под действием раствора гидроксида натрия. Аналогично определяют бигумаль. Препараты бензилпенициллина осаждают в виде N-этилпиперидиновой соли бензилпенициллина; прогестерон — в виде гидра- зона. Возможно применение гравиметрии для определения алкалоидов (взвешиванием свободных от примесей оснований или пикратов, пикролонатов, кремневольфраматов, тетрафенилборатов), а также для определения некоторых витаминов, которые осаждают в виде нерастворимых в воде продуктов гидролиза (викасол, рутин) или в виде кремневольфрамата (тиамина бромид). Известны также гравиметрические методики, основанные на осаждении из натриевых солей кислотных форм барбитуратов.
3.3 Титриметрические (объемные) методыНаибольшее применение получил титриметрическии метод. Название происходит от слова "титр" (фр.) — концентрация. Основная операция метода—титрование, заключающаяся в постепенном приливании к раствору анализируемого вещества титрованного раствора до точки эквивалентности. По измеренному объему титрованного раствора рассчитывают количественное содержание вещества.
Титриметрическии метод анализа получил широкое распространение потому, что он позволяет использовать разнообразные химические реакции и определять вещества, учитывая их свойства и строение. Он выполняется быстро, с большой степенью точности, не нуждается в сложном оснащении и может использоваться как в лабораториях, так и в аптеках.
Для количественного определения лекарственного вещества титриметрическим методом необходимы титрованный (стандартный) раствор, набор простой лабораторной посуды (бюретки, пипетки, мерные колбы колбы для титрования) и средств фиксации точки эквивалентности (конечной точки титрования). Последнюю фиксируют как с помощью индикаторов, так и с помощью физико- химических методов, измеряя приборами физическую константу системы (потенциометрическое, амперометрическое титрование и др. способы). Однако не всякая химическая реакция может быть применима для процесса титрования. К реакциям, используемым в титриметрическом методе, предъявляются следующие требования:
1. возможность фиксировать точку эквивалентности (конечную точку титрования);
2. количественное протекание реакции, т. е. в реакцию должно вступить 100 % анализируемого вещества. Для этого необходимо строго соблюдать определенные условия титрования:
3. реакция должна протекать быстро;
4. не допускаются побочные реакции.
В зависимости от типа реакции, положенной в основу титрования, различают;
· кислотно-основное титрование;
· осадительное титрование;
· комплексиметрическое титрование;
· комплексонометрическое титрование;
· окислительно-восстановительное титрование.
Кислотно-основное титрование осуществляется в воде и в неводных средах. Данный метод используется в 40 процентах методик, применяющихся для анализа лекарственных веществ. Им определяют концентрацию кислот, оснований, солей. В основе титрования лежит реакция взаимодействия протонов с гидроксид-ионами: НзО+ + ОН- = 2Н2О. Титрованными (стандартными растворами являются растворы сильных кислот и сильных оснований. В процессе титрования изменяется рН системы. В зависимости от свойств определяемого вещества точка эквивалентности при титровании в воде может соответствовать различным величинам рН: Очевидно важно подобрать индикатор таким образом, чтобы величина рН в точке эквивалентности находилась в интервале перехода окраски выбранного индикатора.
В качестве индикаторов служат красители, изменяющие окраску в широком интервале рН от 1,2 до 10,5. Наиболее часто используются индикаторы: метиловый оранжевый (3,1—4,4); метиловый красный (4,8—6,0); фенолфталеин (8,2—10,0); тимол-фталеин (9,4—10,6).
Пример подбора индикатора. Подобрать индикатор для определения концентрации бензойной кислоты С6Н5СООН.
Решение. В процессе титрования бензойной кислоты (рКа=4,20) раствором гидроксида натрия образуется соль бензоат натрия
С6Н5СООН+NаОН = С6Н5СOONа+Н20
Бензоат натрия в воде подвергается процессам диссоциации и гидролиза:
С6Н5СООNа -> С6Н5СОО- + Nа+;
С6Н5СОО- + Н20 = С6Н5СООН + ОН-.
В растворе накапливается определенное количество ОН-, их концентрация превысит концентрацию протонов, и поэтому величина рН будет более 7. Это подтверждается приведенными ниже расчетами по формуле рН для растворов солей слабых кислот и сильных оснований: рН = 7 + ½*рКа + ½*lgСсоли, где рКа = 4,20 (табличная величина), а Ссоли определяют, ориентируясь на концентрацию титрованного раствора. Если титруют 0,1 М раствором гидроксида натрия, то Ссоли = 0,1 моль/л. В этом случае lgС = lg0,1 и lgС = -1; ½*0,1 = -0,5. Подставив значения рКа и ½*lgС в рН = 7 + ½*4,20— 0,5=8,6, найдем значение рН в точке эквивалентности. Эта величина находится в интервале рН для фенолфталеина (8,2 — 10,0), следовательно, бензойную кислоту нужно титровать с индикатором фенолфталеином.
Значительное количество лекарственных веществ проявляет способность отщеплять или присоединять протоны и согласно современным теориям являться кислотами или основаниями. Мерой кислотности вещества служит величина показателя кислотности рКа = -1gКа, где Ка— константа ионизации. Чем меньше величина рКа, тем сильнее кислота, тем легче отщепляются протоны. Аналогично рКв — показатель основности вещества. Чем меньше величина рКв, тем сильнее основание, тем активнее вещество присоединяет протоны. Значения рКа и рКв для одного и того же вещества в разных растворителях различны, и этот фактор используют для выбора условий титрования.
ГФ XI приводит значения рКа для ряда лекарственных веществ в различных растворителях. Зная величину рКа, можно решить вопрос о возможности и условиях титрования вещества. Например, для соляной кислоты в воде рКа=0,8; для уксусной кислоты рКа=4,75; для ацетилсалициловой кислоты рКа=3,50. Эти кислоты можно титровать в воде раствором гидроксида натрия. Если величина рКа больше восьми единиц рН, то водная среда не подходит. Например, для титрования барбитала (рКа=7,47), фенола (рКа=9,89), борной кислоты (рКа=9,24) требуются особые условия. Барбитал титруют в среде диметилформамида бензольно-метанольным раствором гидроксида натрия. Борную кислоту превращают добавлением глицерина в диглицеринборную кислоту, которая является более сильной кислотой.
Свои основные свойства в водных и спиртовых средах проявляют лекарственные вещества, присоединяющие протон. Это—амидопирин (рКв=9,2), гексаметилентетрамин (рКв=9,1), алкалоиды, например кодеин (рКв=6,0),. поэтому их можно титровать раствором сильной кислоты.
В водной среде кислотами титруют натриевые соли слабых кислот, так как в их растворе вследствие гидролиза образуется щелочная среда. Соли алкалоидов, в водных растворах которых возникает кислая среда вследствие гидролиза, титруют раствором гидроксида натрия. В процессе титрования солей образуются кислоты или основания, присутствие их оказывает существенное влияние на рН раствора, поэтому их удаляют путем экстрагирования растворителями, не смешивающимися с водой. Например, салицилат натрия, бензоат натрия титруют в присутствии эфира. А соли алкалоидов в присутствии спиртово-хлороформной смеси (1:1). Для алкалиметрического определения аминокислот используется метод формольного титрования (титрование по Серенсену). Наличие аминогруппы, способной присоединять протоны, и карбоксильной группы, отдающей протоны, приводит к тому, что в водных растворах аминокислоты существуют в виде диполярных ионов +NH3-R-СОО-, поэтому полностью оттитровать такие вещества раствором гидроксида натрия не удается. Во избежание этого в раствор перед титрованием добавляют нейтрализованный формалин. Образуется ТЧ-метиленовое производное и устраняется влияние аминогруппы
Если вещество — очень слабая кислота с рКа > 9, например теофиллин (рКа=11,40), его непосредственно оттитровать нельзя. В таком случае прибегают к заместительному титрованию, сущность которого заключается в том, что к раствору анализируемого вещества добавляют несколько капель раствора нитрата серебра. Выделяющееся эквивалентное количество азотной кислоты определяют, алкалиметрически:
Титрование в неводных средах имеет преимущество перед водным титрованием потому, что позволяет определять концентрацию слабых кислот и оснований, часто мало растворимых в воде. Этот метод позволяет также определять соли слабых кислот и слабых оснований, которые невозможно оттитровать в воде. Удобен метод и для анализа многокомпонентных смесей,, часто без их предварительного разделения. Метод позволяет определять физиологически активную часть в солях алкалоидов.
Метод неводного титрования дает более точные результаты по сравнению с титрованием в воде, так как вследствие небольшого поверхностного натяжения неводных растворителей размеры капель титрованных растворов меньше капель водных растворов.
В теории неводного титрования большую роль играет влияние растворителя, на кислотно-основные свойства анализируемого вещества. Для неводного титрования применяются различные растворители, которые по своим свойствам делятся на четыре группы.
Основные (протофильные) растворители легко присоединяют протоны, усиливают кислотные свойства титруемых веществ. Среди них — диметилформамид НСОN(СН3)2, пиридин, жидкий аммиак и др. В среде основных растворителей легко титруются слабые кислоты, кислые формы .барбитуратов, сульфаниламидов, фенолы. Кислотность этих соединений в среде данных растворителей повышается, и тем самым улучшается процесс и результаты титрования. Титрантом служит раствор гидроксида натрия в смеси метанола и бензола или раствор метилата натрия. В качестве индикатора применяют тимоловый синий. Например, при титровании барбитала в среде диметилформамида раствором гидроксида натрия происходят следующие процессы:
2. Кислотные (протогенные) растворители: муравьиная кислота НСООН, уксусная кислота СН3СООН (безводная), уксусный ангидрид и др. Они легко отдают протоны, усиливая основные свойства веществ. Титрантом служит раствор хлорной кислоты, а индикатором — раствор кристаллического фиолетового, тропеолина 00 или метилового оранжевого. Растворы титранта и индикатора готовят в безводной уксусной кислоте. Суммарно процесс нейтрализации слабого органического основания хлорной кислотой представлен следующей схемой:
R3N + HClO4 → [R2N • H+]ClO4-
Подобно происходит титрование производные пиридина (никотинамид, фтивазид), алкалоидов, представляющих собой слабые основания.
Амфотерные (амфипротные) растворители: вода Н2О, этанол С2Н5ОН, метанол СН3ОН и др. Эти растворители могут отдавать свои или присоединять протоны от титруемых веществ. В амфипротных растворителях титруют смеси различных кислот.
Индифферентные (апротонные) растворители: углеводороды — бензол и его производные, галоген-производные углеводородов (хлороформ, четыреххлористый углерод и др.). Молекулы этих растворителей не способны ни отдавать, ни присоединять протоны. В них титруются смеси оснований.
Недостатком неводного титрования является необходимость иметь герметизированную титровальную установку. Работа предполагает использование токсичных, летучих растворителей. Однако метод позволяет определять концентрацию солей слабых кислот и слабых оснований, что не всегда возможно в водной среде. Ацетат калия титруется хлорной кислотой по схеме
СН3СООК + HClO4 →KClO4 + СН3СООН.
Титрование солей слабых оснований (R3N • HA) •можно выразить следующей схемой
R3N • HA + HClO4 → [R3N • H]ClO4 + HA.
Так титруются адреналин и норадреналин гидротартраты, нафтизин, цитрат дитразина, соли алкалоидов (фосфат кодеина, гидротартрат платифиллина, сульфат атропина, бензоат сферофизина). Однако соли галогенводородных кислот (гидрохлориды, гидробромиды, гидроиодиды) алкалоидов и азотсодержащих оснований не могут быть непосредственно оттитрованы хлорной кислотой, так как галоген-ионы проявляют кислотные свойства даже в среде безводной уксусной кислоты и поэтому могут влиять на переход цвета индикатора в точке эквивалентности. Титрование солей галогенводородных кислот выполняют в присутствии ацетата ртути (II), который связывает галоген-ионы в малодиссоциированные соединения (дихлорид, дибромид или дийодид ртути), и титрование идет с хорошими результатами по схеме
2R3N • HX + Hg(CH3COO)2 → HgX2 + 2[R3NH]+CH3COO-.
2[R3NH]+CH3COO- + 2HClO4 → 2[R3NH]+ClO4 + 2CH3COOH.
Возможность и оптимальные условия титрования в неводных средах определяются величиной константы титрования Кт, которую рассчитывают, исходя из величин ионного произведения среды Кi и Ка — константы диссоциации титруемого вещества в этой среде, по формулам для кислот Кт = Кi/Ка, для оснований Кт=Ка или рКт=рК; -рКа и рКт=рКа. Чем меньше числовое значение Кт и чем больше рКт, тем условия титрования лучше. Значения величины Кi; и рКi; для ряда растворителей и Ка, а также рКа для некоторых лекарственных веществ приведены в ГФ XI.
Пример выбора среды для количественного определения ацетилсалициловой кислоты.
Величина рКi этанола 19,1; воды — 14. Для ацетил-салициловой кислоты рКа=3,50. В воде рКт=14-3,50=10,5; в этаноле рКт=19,1-3,50=15,6.
Величина рКт в этаноле больше, следовательно в этом растворителе условия титрования ацетилсалициловой кислоты лучше.
В ряде случаев для титрования применяют смеси неводных растворителей с апротонными растворителями: бензолом, хлороформом и др., присутствие которых уменьшает ионное произведение среды К; что способствует улучшению условий титрования.
Осадительное титрование. В основу метода положе на реакция образования малорастворимого соединения, В фармацевтическом анализе широко используют аргентометрию, которая предполагает взаимодействие галогенов с нитратом серебра:
МеНаl + АgNО3 → АgНаl↓ + МеNO3.
Применяется метод в виде прямого (методы Мора, Фаянса) и обратного титрования (метод Фольгарда) Титрантами являются 0,1 М и 0,05 М растворы нитрат серебра и тиоцианата аммония.
По методу Мора титрование раствором нитрата серебра выполняют при рН 6,5—10,0 в присутствие 5—7 капель 5 %-ного водного раствора хромата калия в качестве индикатора. В процессе титрования образуются малорастворимые галогениды серебра, и, когда и осаждение закончится полностью, образуется красный осадок хромата серебра, свидетельствующий о достижении точки эквивалентности:
К2СrО4, + 2АgNO3 → Ag2СrО4↓ + 2КNO3
Этим методом определяют концентрацию хлоридов и бромидов. Иодиды определять не рекомендуется, потому что появление красной окраски происходит ранее точки эквивалентности, что объясняется адсорбцией иодид-ионов поверхностью осадка.
Метод Фаянса применяется для определения концентрации йодидов, но он может использоваться также для хлоридов и бромидов. В отличие от метода Мора, титрование выполняется не только в нейтральной среде, но и в среде уксусной кислоты с водным раствором эозината натрия в качестве индикатора. В точке эквивалентности наблюдается появление ярко-розового окрашинивания осадка. Хлориды и бромиды титруют в среде уксусной кислоты, индикатором служит раствор бром-фенолового синего. В точке эквивалентности зеленовато-желтое окрашивание переходит в сине- фиолетовое. Метод Фольгарда используется для определения концентрации хлоридов, бромидов, йодидов способом обратного титрования. Индикатором является раствор железоаммониевых квасцов. Анализ производится в среде азотной кислоты.К отобранному для определения концентрации раствору приливают точно измеренный избыток раствора нитрата серебра, 2—3 мл разведенной азотной кислоты, 10 капель раствора железоаммониевых квасцов и титруют раствором тиоцианата аммония до появления розовой окраски:
АgNО3 + NН4SCN → AgSСN↓ + NH4NO3,
3NН4SCN + FeNH4(SO4)2 → [Fe(SCN)3] + 2(NH4)2SO4
При титровании хлоридов возможно взаимодействие осадка хлорида серебра с красным соединением:
[Fe(SCN)3] + 3АgСl → 3АgSСN↓ + FеС13
и определение точки эквивалентности затрудняется. Чтобы избежать протекания реакции между хлоридом серебра и комплексным соединением, можно отфильтровать осадок и в фильтрате оттитровать избыток нитрата серебра. Но можно перед титрованием в анализируемый раствор добавить 5—10 мл органического растворителя с большой плотностью, например четыреххлористого углерода, который покрывает поверхность осадка хлорида серебра, и тогда взаимодействие осадка не происходит. При титровании йодидов индикатор — раствор железоаммониевых квасцов — прибавляют после добавления избытка нитрата серебра. Если этого не сделать, то возможно окислительно-восстановительное взаимодействие йодид-иона с индикатором
2КI + 2 FeNH4(SO4)2 → 2FeSO4 + I2 + (NН4)2SO4 + К2SO4.
Видоизмененный метод Фольгарда используется при определении хлоридов и йодидов. Этот способ позволяет избежать взаимодействия йодидов с железоаммониевыми квасцами и осадка хлорида серебра с комплексным соединением [Fe(SCN)3] и тем самым улучшить условия титрования.
К растворенной навеске галогенида добавляют 2—3 мл разведенной азотной кислоты, 1 мл раствора железоаммониевых квасцов и 0,1 мл 0,1 М раствора тиоцианата аммония. Раствор окрашивается в красный цвет. Титруют 0,1 М раствором нитрата серебра до исчезновения окраски. При расчетах необходимо учитывать объем раствора нитрата серебра, который затратится на 0,1 мл 0,1 М раствора тиоцианата аммония, добавленного в анализируемый раствор до начала титрования.
Описанные выше методы осадительного титрования не являются избирательными, при анализе раствора смеси галогенидов определяется их общее содержание. Для определения йодидов в растворах с хлоридами и бромидами существуют избирательные методы.
Метод Кольтгофа является методом прямого аргентометрического титрования раствором нитрата серебра. К анализируемому раствору приливают 3 мл воды, 3 мл 10%-ного раствора карбоната аммония, 3—4 капли разведенной серной кислоты, 5—6 капель раствора крахмала, одну каплю 0,1 М раствора йодата калия. Раствор окрашивается в синий цвет вследствие выделения йода согласно уравнению
5KI + КIO3 + 3H2SO4 → 3I2 + 3K2SO4 + 3Н20.
Раствор медленно, тщательно перемешивая, титруют раствором нитрата серебра до перехода синей окраски в желтую, обусловленную цветом осадка йодида серебра. В процессе титрования в растворе уменьшается концентрация йодида калия, равновесие смещается влево, уменьшается концентрация йода, и синяя окраска исчезает Возможность определения йодидов в присутствии остальных галогенидов достигается потому, что в растворе образуется буферная смесь, поддерживающая значение рН<5,5. Бромиды в этих условиях не окисляются йодатом калия при его незначительной концентрации.
Другим методом прямого аргентометрического титрования является метод Шика. К анализируемому раствору приливают 4—5 мл воды, 5 мл разведенной серной кислоты и титруют раствором нитрата серебра. Индикатор — нитрозокрахмальная бумага (полоска фильтровальной бумаги, пропитанная смесью растворов нитрита натрия и крахмала). При нанесении на ее поверхность капли раствора до момента эквивалентности бумага окрашивается в синий цвет вследствие протекания реакции
2NaNO2 + 2NaI + 2H2SO4 → I2 + 2Na2SO4 + 2H20 + 2NO
Цвет бумаги не изменится после достижения точки эквивалентности. Для получения более точных результатов целесообразно предварительно рассчитать количество титрованного раствора нитрата серебра, необходимого для титрования взятого количества йодидов, или выполнить ориентировочное титрование, а затем при повторном титровании иметь в виду результаты расчетов.
Несколько видоизмененный метод Фольгарда нашел применение не только для определения содержания галогенид-ионов. В его основе лежит способность некоторых органических веществ осаждаться солями серебра.
Например, концентрацию никотиновой кислоты можно определить, после нейтрализации ее аликвотной части, осаждением точно измеренным избытком титрованного раствора нитрата серебра. Происходит образование малорастворимой соли. Через 30 мин осадок отфильтровывают. К аликвотной части фильтрата прибавляют несколько капель разведенной азотной кислоты, раствор железоаммониевых квасцов и титруют 0,1 М раствором тиоцианата аммония до появления розового окрашивания раствора.
Концентрацию дибазола выявляют следующим образом: вначале его осаждают в виде малорастворимой соли серебра, затем осадок с фильтром переносят в колбу для титрования, прибавляют 1—2 мл разведенной азотной кислоты, слегка подогревают до растворения осадка. Охлажденный раствор титруют раствором тиоцианата аммония, используя в качестве индикатора железоаммониевые квасцы до появления розовой окраски раствора.
Для определения содержания сульфатов титрантами служат 0,1 М растворы солей нитрата свинца и нитрата бария. С целью создания оптимальных условий титрования в анализируемый раствор добавляют ацетон или 95%-ный спирт.
Сульфат атропина определяют титрованием раствором нитрата сйинца с индикатором дифенилкарбазидом до перехода желтого окрашивания в розовое. Концентрацию некоторых сульфатов можно выявлять титрованием раствором нитрата бария в присутствии в качестве индикатора 0,2%-ного водного раствора карбоксиарсеназа. Так определяют сульфат атропина, сульфат стрептомицина, сульфат цинка, сульфат хинина.
Комплексиметрический метод основан на образовании комплексного соединения. Меркуриметрия используется для определения концентрации галогенидов, тиоцианатов, цианидов с помощью титрованного раствора — нитрата ртути (II). Предложен также раствор перхлората.ртути (II). Титрованные растворы готовят с добавлением соответствующих кислот. Точку эквивалентности устанавливают потенциометрически или с помощью индикатора дифенилкарбазида, образующего с избытком соли ртути (II) сине-фиолетовое соединение:

При определении йодидов в процессе титрования образуется бесцветное комплексное соединение 4КI + Hg(NO3)2= К2[НgI4] + 2КNO3.
Точку эквивалентности определяют по образованию неисчезающего красного осадка дийодида ртути (II) К2[HgI4] + Нg(NO3)2 = 2HgI2 + 2КNО3.
Иодиды можно титровать с индикатором дифенилкарбазидом, если в титруемый раствор добавить несколько миллилитров этанола. Красный осадок дийодида ртути (II) растворяется в этаноле, и тогда точку эквивалентности определяют с индикатором по появлению сине-фиолетового окрашивания. Титрование выполняется в кислой среде.
При работе с солями ртути (II) необходимо соблюдать осторожность, помнить, что растворимые соли ртути ядовиты.
К комплексиметрическому титрованию относится куприметрическое определение левомицетина. К раствору левомицетина добавляют несколько капель раствора гидроксида натрия, мурексид (как индикатор) и медленно приливают титрованный раствор сульфата меди до изменения окраски раствора из фиолетовой в коричнево-красную, сравнивая ее с окраской контрольного раствора.
При добавлении гидроксида натрия к раствору левомицетина происходит взаимодействие
В процессе титрования образуется комплексное соединение:
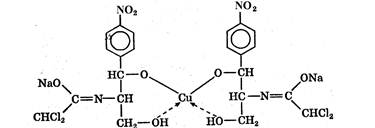
Комплексонометрический метод основан на реакции образования прочных комплексов полиаминокарбоновых кислот с ионами металлов (Са2+, Mg2+,Zn2+, Вi2+ и др.). Наиболее широко применяется динатриевая соль этилендиаминтетрауксусной кислоты (трилон Б). Трилон Б наряду с карбоксильными группами содержит аминный азот. Вследствие такого строения он является кислотой и комплексообразующим веществом. Многие металлы заменяют атомы водорода карбоксильных групп, одновременно связываясь координационно с азотом аминогруппы и образуя прочные комплексы трилона Б с металлом. Двухзарядный катион (например, Са2+) образует комплексное соединение следующего состава:
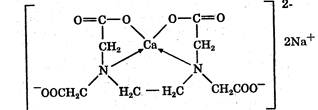
Образование комплексов можно представить схематично:
Na2H2I → 2Na+ + H2I2-
Ме2+ + H2I2- → MeI2- + 2Н+
где Na2H2I — трилон Б; Ме2+ — ион металла. Как видно из приведенной схемы, реакция образования комплексов сопровождается накоплением протонов в растворе, поэтому связывание Н+- ионов должно способствовать образованию комплекса. Наиболее благоприятный для комплексообразования реакцией среды является рН 8—10. Поэтому титрование солей металлов трилоном Б проводят в присутствии аммиачного буфера. Для установления точки эквивалентности применяются специальные индикаторы, которые являются органическими красителями. К ним относятся кислотный хром темно-синий, кислотный хром черный специальный, называемый эриохром черный Т, мурексид, калькон-карбоновая кислота и др. Процесс комплексонометрического титрования заключается в том, что к исследуемому раствору, содержащему определяемый катион, при строго определенном значении рН прибавляют индикатор, при этом образуется хорошо растворимое в воде окрашенное комплексное соединение индикатора с ионом определяемого металла. При титровании трилоном Б этот комплекс разрушается и образуется более прочный, как правило бесцветный, комплекс иона металла с трилоном Б. При этом выделяется анион индикатора, который окрашивает раствор в цвет, присущий свободному индикатору при данном значении рН:
Са2+ + Н2Ind- → CaInd- + 2Н+
CaInd- + H2I2- → CaI2- + H2Ind-
Комплексонометрическое титрование осуществляется как методом прямого, так и методом обратного титрования. Оно позволяет определять количественное содержание солей, оксидов металлов магния, кальция, цинка, свинца, висмута, ртути и др. Метод пригоден также для раздельного определения солей металлов в смеси. Раздельное определение солей кальция и магния при их совместном присутствии основано на том, что растворимость соединений титруемых солей зависит от величины рН в анализируемом растворе. Аликвотную часть раствора титруют вначале с индикатором эриохром черным при рН = 9 в присутствии аммиачного буферного раствора, причем титруются обе соли. В другой аликвотной части определяют соль кальция. В раствор добавляют несколько миллилитров 20%-ного раствора гидроксида натрия, рН этого раствора изменяется от 9 до 12. В этих условиях соли магния осаждаются в виде гидроксида магния, и далее титруют соль кальция с индикатором мурексидом.
Интерес представляет косвенное комплексонометрическое определение аминопроизводных и солей органических оснований (гидрохлорида папаверина, прозерина, спазмолитина, производных фенотиазина). В этом случае используется раствор тетрароданоцинкат (П)-аммония (ТРЦ), который получают при взаимодействии тиоцианата аммония и сульфата цинка:
Для приготовления 0,5 М раствора ТРЦ берут 144 г сульфата цинка, помещают в мерную колбу вместимостью 1 л, добавляют 152 г тиоцианата аммония, растворяют в воде и доводят водой до 1 л. Тщательно перемешивают, фильтруют через вату, хранят при комнатной температуре. При хранении реактива может выпасть осадок или измениться цвет. Несмотря на эти изменения, реактив годен к применению.
4NH4SCN + ZnSO4 → (NH4)2[Zn(SCN)4] + (NH4)2SO4
При добавлении реактива ТРЦ к анализируемому раствору образуется осадок комплексной соли. Например, при определении прозерина происходит осаждение его в соответствии с уравнением реакции

Осадок экстрагируют точно измеренным объемом хлороформа при энергичном взбалтывании в течение 2 мин и фильтруют через бумажный фильтр в сухую колбу. В колбу для титрования отбирают пипеткой определенный объем фильтрата, приливают избыток титрованного раствора трилона Б. Часть титранта вступает во взаимодействие с цинком, образуя прочное комплексное соединение. Не вошедший в реакцию титрант в присутствии аммиачного буферного раствора и индикатора хром темно-синего оттитровывают раствором сульфата цинка.
Окислительно-восстановительное титрование основано на окислительных или восстановительных свойствах анализируемых веществ и титрантов. В процессе титрования происходит изменение окислительно-восстановительных потенциалов взаимодействующих систем. Если разность этих потенциалов достаточно большая, то окислительно-восстановительный процесс протекает практически до конца, и поэтому возможно прямое титрование.
В фармацевтическом анализе применяют такие методы окислительно-восстановительного титрования, как перманганатометрия, йодометрия, йодхлорометрия, йодатометрия, броматометрия, дихроматометрия, цериметрия.
ПЕРМАНГАНАТОМЕТРИЯ основана на использовании окислительных свойств титранта — перманганата калия в кислой и щелочной средах.
МnО4- + 8Н+ + 5е → Мn2+ + 4Н20.
В кислой среде продуктом восстановления являются практически бесцветные соли марганца (II). Поэтому при прямом перманганатометрическом титровании индикатор в анализируемый раствор не добавляют, им является титрант, избыток которого придает раствору розовое окрашивание.
Прямое титрование используется для определения восстановленного железа, препаратов пероксида водорода.
В случае медленного протекания окислительно-восстановительного процесса применяется обратное титрование. Например, при определении нитрита натрия, левомицетина. Избыток титранта оттитровывают йодометрически.
ИОДОМЕТРИЯ основана на использовании окислительных свойств свободного йода и восстановительных свойств йодид-ионов:
I2 + 2е → 2I-
Методом йодометрии количественно определяют неорганические и органические лекарственные вещества, способные окисляться или восстанавливаться, а также образовывать с йодом продукты замещения. Кроме того, йодометрию используют для определения избытка титранта в обратном окислительно-восстановительном титровании. Свободный йод, образовавшийся в избытке при обратном йодометрическом титровании, оттитровывают тиосульфатом натрия:
I2 + 2Nа2S2О3 → 2NаI + Nа2S4О6.
Индикатором обычно служит раствор крахмала, образующий с йодом синее соединение. Прямое титрование йодом применяют для определения тиосульфата натрия и препаратов мышьяка (III). Иодометрическое опреде ление, основанное на окислении альдегидов йодом, используют для титрования хлоралгидрата, формальдегида в растворе, а также лекарственных веществ образующих формальдегид при гидролизе (никодина метазида). Окисление альдегидов происходит по схеме:
I2 + 2NаОН → NaIO + NaI + Н2O
Процесс окисления йодом лежит в основе определении фурацилина, изониазида, метионина, анальгина и др.
Восстановительные свойства йодида калия используются для определения окислителей. Лекарственные вещества — окислители выделяют эквивалентное количество свободного йода при взаимодействии с йодидом калия. Выделившийся йод титруют раствором тиосульфата натрия. Эти процессы лежат в основе колчественного определения препаратов пероксида водорода, соединений мышьяка (V), меди (II), перманганата калия, а также обладающих окислительными свойствами гипохлоридов (известь хлорная) и хлор-производных амидов сульфокислот (хлорамины, пантоцид). Для количественного анализа используется сочетание реакций замещения и йодометрии. С помощью титрованного раствора йода получают йодпроизводные, их отфильтровывают, а в фильтрате определяют избыток йода титрованием раствором тиосульфата натрия. Этот прием используют для определения некоторых алкалоидов, которые образуют малорастворимые перйодиды состава [R3N]•НI•I4. Это кодеин, кокаин, кофеин и др.
ИОДХЛОРОМЕТРИЮ согласно ГФ XI рекомендуют для количественного определения лактата этакридина, который осаждается в виде дийодпроизводного. Избыток титранта раствора йодмонохлорида оттитровывают йодометрически:
IСl + КI →I2 + КСl.
Иодхлорометрическим методом можно определять фенолы, сульфаниламиды и другие первичные ароматические амины.
ИОДАТОМЕТРИЮ используют для определения фтивазида, апрессина, аскорбиновой кислоты. Происходит процесс окисления лекарственных веществ титрованным раствором йодата калия КIO3. Например, по ГФ X аскорбиновую кислоту рекомендуют титровать 0,1 М раствором йодата калия в присутствии йодида калия. Окисление происходит по схеме:
Избыток титранта йодата калия в точке эквивалентности приводит к окислению йодида калия в кислой среде в соответствии с уравнением
КIO3 + 5KI + 6НСl → 3I2 + 6KCl + 3Н20.
Иод окрашивает раствор в желтый цвет, а после добавления раствора крахмала — в синий цвет. Фтивазид титруют раствором йодата калия после предварительного гидролиза в среде соляной кислоты. В титруемый раствор прибавляют несколько миллилитров хлороформа. Образующийся при титровании йод извлекается хлороформом, окрашивая его в фиолетовый цвет: Точку эквивалентности определяют по обесцвечиванию хлороформного слоя, когда йод превратится в монохлорид иода.
В БРОМАТОМЕТРИИ в качестве титранта применяется раствор бромата калия. Титрование восстановителей, таких, как мышьяк (III), сурьма (III), гидроксиламин, производные гидразина и др., можно осуществлять прямым и обратным титрованием в среде соляной и серной кислот. Бромат калия восстанавливается до бромида калия, и в тот момент, когда в растворе появляется небольшой избыток титранта, тотчас реагирует с ним:
КBrО3 + 5KBr + 6НС1 → 3Br2 + 6КС1 + 3Н20.
Образовавшийся свободный бром окрашивает раствор в бледно-желтый цвет. Однако эта окраска очень слабая и точку эквивалентности по ней точно фиксировать нельзя, поэтому пользуются индикаторами метиловым оранжевым и метиловым красным. В точке эквивалентности индикатор необратимо окисляется до бесцветных продуктов.
Броматометрические определения основаны не только на окислительно-восстановительных процессах, но и на реакциях присоединения брома и замещения бромом, который образуется в процессе взаимодействия с бромидом калия. Поэтому очень часто титрование производят титрованным раствором, содержащим бромат калия и бромид калия в соотношении 1:5. При использовании метода обратного титрования избыток титранта определяют йодометрически. Метод используют для определения лекарственных веществ производных фенолов (фенол, тимол, резорцин, салициловая кислота) и первичных ароматических аминов (сульфаниламидные препараты, производные п-аминобензойной кислоты), йодидов и органических оснований.
ДИХРОМАТОМЕТРИЯ использует в качестве титранта раствор дихромата калия. Титрование выполняется в среде серной, соляной или фосфорной кислот.
Применение этого метода основано на реакциях окисления-восстановления и реакциях двойного обмена, сопровождающихся образованием не растворимых в воде соединений. Так определяют концентрацию метиленового синего, акрихина, этония. К растворенной навеске анализируемого вещества приливают избыток титрованного раствора, происходит образование осадка дихромата основания:
2 (R3N • Н)+Сl- + К2Сr2О7 → [R3N • Н]2Сг2O7↓ + 2КС1.
После отделения осадка фильтрованием избыток титранта в фильтрате определяют йодометрическим методом:
К2Сr2О7 + 6KI + 7H2SO4 → Cr2(SO4)3 + 3I2 + 4K2SO4 + 7Н20.
Выделившийся йод титруют раствором тиосульфата натрия.
ЦЕРИМЕТРИЯ основана на применении титрованного раствора солей церия (IV), которые в кислой среде восстанавливаются до церия (III)
Се4+ + е = Се3+
Соединения церия (IV) обладают устойчивостью титрованных растворов как при комнатной температуре, так и при нагревании до 100°С и выше.
Метод предложен для определения содержания неорганических соединений, таких, как железо (II), мышьяк (III), и органических лекарственных веществ (углеводов, органических кислот, викасола, производных фенотиазина).
НИТРИТОМЕТРИЯ использует в качестве титранта раствор нитрита натрия. Метод применяется главным образом для определения органических лекарственных веществ. Наибольшее количество методик основано на легко протекающих реакциях диазотирования или нитрозирования. Первичные ароматические амины образуют с нитритом натрия в среде соляной кислоты диазосоединение
Ar—NH2 + NaNO2 + 2HCl → [Ar—N+≡N)C1- + NaCl + 2H20.
Вторичные ароматические амины в таких же условиях образуют нитрозоамины
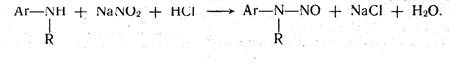
Эквивалентную точку устанавливают с помощью внешних и внутренних индикаторов, а также потенциометрическим методом. Реакция диазотирования является экзотермической и к концу, титрования протекает медленно. Для ускорения в анализируемый раствор прибавляют катализатор — кристаллический бромид калия.
Область применения нитритометрии—определение сульфаниламидных препаратов, производных п-аминобензоиной кислоты.
Расчеты результатов анализа. Содержание вещества (в граммах) в 100 лекарственной формы вычисляют по формуле
![]()
где V— объем титрованного раствора; КП — поправочный коэффициент; Т — титр раствора по определяемому веществу, г; а — масса или объем вещества, взятого для анализа.
Содержание вещества (в граммах) во всей массе или во всем объеме лекарственной формы определяют по формуле
![]()
где Р — масса или объем лекарственной формы.
Титр раствора по определяемому веществу показывает массу определяемого вещества, которое реагирует с миллилитром титрованного раствора, рассчитывается по формуле
![]()
где N—нормальность титрованного раствора; E— величина одного моля эквивалента определяемого вещества.
3.4 Газометрический анализГазометрический метод имеет ограниченное применение в фармацевтическом анализе. Объектами газометрического анализа являются два газообразных препарата: кислород и циклопропан. Сущность газометрического определения кислорода заключается во взаимодействии с поглотительным раствором, содержащим легко окисляющийся медноаммиачный комплекс. Определение выполняют в приборе Гемпеля (ГФ IX, с. 349), измеряя объем непрореагировавшего газа. Циклопропан определяют аналогично, используя в качестве поглотительного раствора концентрированную серную кислоту (ГФ X, с. 228).
3.5 Количественный элементный анализЭлементный анализ используют для количественного определения органических и элементорганических соединений, содержащих азот, галогены, серу, а также мышьяк, висмут, ртуть, сурьму и другие элементы.
Фармакопейный метод определения азота в органических соединениях известен также под названием метода Кьельдаля. Он основан на сочетании минерализации органического вещества с последующим применением кислотно-основного титрования. Применяют метод Кьельдаля для количественного анализа азотсодержащих органических веществ, а также лекарственных препаратов, содержащих аминный, амидный и гетероциклический азот. Метод включает несколько последовательно выполняемых стадий. Вначале осуществляют минерализацию образца нагреванием с концентрированной серной кислотой.
R-NH2 + [O] + H2SO4 → CO2↑ + H2O + NH4HSO4
Затем действуют на гидросульфат аммония гидроксидом натрия и отгоняют выделяющийся аммиак в приемник, содержащий раствор борной кислоты.
NH4HSO4 + 2NaOH → NH3↑ + 2H2O + Na2SO4
Так как борная кислота реагирует с аммиаком с образованием солей метаборной и тетраборной кислот, то в приемнике образуются метаборат и тетраборат аммония:
NH3 + Н3ВО3 → NH4ВO2 + Н2O
2NH3 + 4Н3ВO3 → (NH4)2В4O7 + 5Н2O
Далее собранный отгон, содержащий весь образовавшийся аммиак в виде мета- и тетрабората аммония, титруют 0,1 М раствором соляной кислоты:
NH4ВO2 + НС1 + H2O → NH4С1 + Н3ВО3
(NH4)2В4O7 + 5Н20 + 2НС1 → 2NH4С1 + 4Н3ВO3
Для повышения точности анализа параллельно выполняют контрольный опыт. Разность между количеством миллилитров титрованного раствора соляной кислоты в основном и контрольном опытах, умноженная на 0,0014, соответствует количеству азота (г), который содержится в испытуемом веществе.
Область применения метода Кьельдаля в фармацевтическом анализе довольно широка. ГФ рекомендует его для определения уретанов (мепротан), аминокислот (метионин, глутаминовая кислота) и других азотсодержащих лекарственных веществ (бензогексоний, оксафенамид, дипрофиллин). Самый существенный недостаток метода — его трудоемкость.
Для определения некоторых лекарственных веществ, содержащих легко гидролизующуюся в щелочной среде амидную группу (салициламид, диэтиламид никотиновой кислоты, салюзид растворимый, прозерин), используют упрощенный вариант метода Кьельдаля, исключающий стадию минерализации. Методика определения сводится к разрушению препарата 30%-ным раствором гидроксида натрия в колбе Кьельдаля и отгонке выделяющегося аммиака (или диалкиламина) в приемник.
Взамен трудоемкого метода определения содержания азота в органических лекарственных веществах (метода Кьельдаля) предложен унифицированный способ, основанный на сочетании высокотемпературной окислительной деструкции в замкнутом объеме с последующим газохроматографическим разделением и определением образовавшегося азота (Н.С.Евтушенко). Способ применен для количественного определения метилурацила, пентоксила, аллацила, тетридина, пиперазина, диазолина, бензогексония, дитразина цитрата, которые по НТД определяют методом Кьельдаля или гравиметрическим методом. Анализ после сожжения навески лекарственного вещества выполняют на газовом хроматографе типа ЛХМ-8МД. В качестве стандартного вещества используют п-нитрофенол или п-нитроанилин. Расчет производят методом сравнения высот пиков азота у стандартного и анализируемого веществ. Относительная погрешность определения при анализе индивидуальных веществ не превышает ±2%, продолжительность выполнения 20—25 мин.
В фармацевтическом анализе автоматизированные элементные С-, H-, N-анализаторы наряду с функциональным анализом могут быть использованы для подтверждения подлинности и количественного определения лекарственного вещества. С этой целью используется автоматизированный N-анализатор для унифицированного анализа азотсодержащих лекарственных веществ. Принцип метода состоит в том, что после твердофазного окисления азотсодержащего лекарственного вещества в статическом режиме элементный азот определяют ГЖХ-методом. Метод может использоваться взамен таких трудоемких методов, как гравиметрия и метод Кьельдаля.
Метод сжигания в колбе с кислородом является одним из перспективных методов количественного элементного анализа. Он включен во многие фармакопеи мира, в том числе Международную и Европейскую, но пока ограниченно используется в отечественном фармацевтическом анализе. Метод основан на разрушении органического вещества сожжением в колбе, наполненной кислородом, растворении образовавшихся продуктов в поглощающей жидкости и последующем определении элементов, находящихся в растворе в виде ионов или молекул. Определение выполняют различными химическими или физико-химическими методами. Метод может быть использован для качественного и количественного определения органических лекарственных веществ, содержащих в молекуле галогены, серу, фосфор, азот и другие элементы. Преимущества метода состоят в быстроте процесса минерализации, занимающего несколько секунд; исключении потерь элемента в процессе минерализации, проходящем в герметически закрытой колбе; возможности унификации применительно к различным группам соединений; высокой чувствительности анализа на заключительной его стадии и широкого сочетания метода на этой стадии с физико-химическими методами. Большие перспективы открывает применение метода сжигания в кислороде для определения примесей тяжелых металлов и других элементов в лекарственных веществах при испытании их на чистоту.
ГФ рекомендует этот метод для определения иода в иодорганических лекарственных веществах. Однако проведенные в последние годы исследования подтверждают возможность его использования для элементного анализа сульфаниламидных препаратов и других, содержащих серу, органических лекарственных препаратов. Весьма широки перспективы применения метода для количественного анализа хлор- и бром-содержащих лекарственных препаратов с последующим использованием меркуриметрического титрования. Важная особенность метода сжигания в кислороде — возможность применения для анализа препаратов в таких лекарственных формах, как таблетки, драже, мази, суппозитории.
Количественный элементный анализ галоген-, мышьяк- и ртутьсодержащих лекарственных веществ может быть выполнен после предварительной минерализации в различных условиях. ГФ рекомендует метод количественного определения йодсодержащих органических лекарственных веществ (биллигноста, тиреодина и др.), основанный на их минерализации и окислении образовавшегося иона йода в йодат-ион. Последний определяют, используя в качестве восстановителя йодид калия по общему принципу йодатометрии:
KIO3 + 5KI + 3Н2SO4 → 3I2 + 3K2SO4 + 3Н2O
Разработаны экспрессные методы окислительной минерализации и определения йод-органических соединений. В качестве окислителей для водорастворимых лекарственных веществ используют перманганат калия, а для нерастворимых в воде — пергидроль в кислой среде. В обоих случаях происходит окисление атомов йода до йодат-ионов, которые затем определяют йодометрическим методом.
В последние годы разработан метод количественного определения галогенсодержащих органических лекарственных веществ, основанный на восстановительной минерализации в среде высококипящего растворителя. Образовавшиеся галоген-ионы титруют меркуриметрическим методом, используя в качестве титранта раствор перхлората ртути.
Для анализа органических веществ, содержащих галоген в алифатическом радикале, предложен способ минерализации, который основан на восстановлении галогена калиевой солью муравьиной кислоты в момент ее выдачения при взаимодействии диметилформамида с гидроксидом калия.
Количественный элементный анализ мышьяксодержащих органических лекарственных веществ (осарсол) основан на минерализации органической части молекулы в присутствии окислителей (смеси концентрированных азотной и серной кислот). В результате минерализации образуется смесь ионов мышьяка (III) и мышьяка (V). Содержащиеся в смеси соединения мышьяка (V) при нагревании с сульфатом гидразина восстанавливаются до мышьяка (III):
2H3AsO4 + H2N-NH2 → N2↑ + 2H3AsO3 + 2Н2O
Мышьяк (III) определяют броматометрическим методом.
Ртутьорганическое лекарственное вещество количественно превращают в дихлорид ртути нагреванием на кипящей водяной бане с 0,1 М раствором соляной кислоты. Затем дихлорид ртути действием избытка иодида калия превращают в водорастворимую комплексную соль:
HgCl2 + 2KI → Hgl2↓ + 2КС1
Hgl2 + 2KI —> K2[HgI4]
а избыток кислоты оттитровывают щелочью.
Глава 4. Физико-химические методы анализа 4.1 Особенности физико-химических методов анализа
Физико-химические методы приобретают все большее значение для целей объективной идентификации и количественного определения лекарственных веществ. Получивший распространение в различных отраслях недеструктивный анализ (без разрушения анализируемого объекта) играет важную роль и в фармацевтическом анализе. Для его выполнения пригодны многие физико-химические методы, в частности оптические, ЯМР-, ПМР-, УФ- и ИК- спектроскопия, ГЖХ, ВЭЖХ и др.
В фармацевтическом анализе наиболее широко используют физико-химические методы, которые могут быть классифицированы на следующие группы: оптические методы; методы, основанные на поглощении излучения; методы, основанные на испускании излучения; методы, основанные на использовании магнитного поля; электрохимические методы; методы разделения; термические методы.
Большинство перечисленных методов (за исключением оптических, электрохимических и термических) широко применяют для установления химической структуры органических соединений.
Каждое новое издание ГФ является своеобразным отражением перспектив использования физико-химических методов в фармацевтическом анализе. Так ГФ XI по сравнению с ГФ X пополнилась общими статьями и разделами по таким методам, как ГЖХ, ВЭЖХ, ЯМР-спектроскопия, электрофорез, эмиссионная и атомно-абсорбционная пламенная спектрометрия. Остальные статьи, содержащие описание физико-химических методов, существенно переработаны и дополнены с учетом современных достижений в области теории и практики фармацевтического анализа.
Физико-химические методы анализа имеют ряд преимуществ перед классическими химическими методами. Они основаны на использовании как физических, так и химических свойств веществ и в большинстве случаев отличаются экспрессностью, избирательностью, высокой чувствительностью, возможностью унификации и автоматизации.
4.2 Оптические методыК этой группе относятся методы, основанные на определении показателя преломления луча света в растворе испытуемого вещества (рефрактометрия), измерении интерференции света (интерферометрия), способности раствора вещества вращать плоскость поляризованного луча (поляриметрия).
Оптические методы находят все более широкое применение в практике внутриаптечного контроля ввиду экспрессности, минимального расхода анализируемых лекарств.
Рефрактометрия использована для испытания подлинности лекарственных веществ, представляющих собой жидкости (диэтиламид никотиновой кислоты, метилсалицилат, токоферола ацетат), а во внутриаптечном контроле — для анализа лекарственных форм, в том числе двойных и тройных смесей. Применяют также объемно-рефрактометрический анализ и рефрактометрический анализ методом полной и неполной экстракции.
Разработаны различные варианты методик анализа интерферометрическим методом лекарственных препаратов, титрованных растворов, дистиллированной воды.
Поляриметрию применяют для испытания подлинности лекарственных веществ, в молекулах которых имеется асимметрический атом углерода. Среди них большинство препаратов из групп алкалоидов, гормонов, витаминов, антибиотиков, терпенов.
В аналитической химии и фармацевтическом анализе используются рентгенорефрактометрия порошков, спектрополяриметрический анализ, лазерная интерферометрия, дисперсия вращения и круговой дихроизм.
Помимо указанных оптических методов для идентификации индивидуальных лекарственных веществ в фармацевтическом и токсикологическом анализе не теряет своего значения химическая микроскопия. Перспективно применение электронной микроскопии, особенно в фитохимическом анализе. В отличие от оптической микроскопии объект подвергается воздействию пучка электронов высоких энергий. Изображение, образованное рассеянными электронами, наблюдают на флуоресцирующем экране.
Одним из перспективных экспрессных физических методов является рентгенографический анализ. Он позволяет идентифицировать лекарственные вещества в кристаллической форме и различать при этом их полиморфное состояние. Для анализа кристаллических лекарственных веществ могут быть также применены различные виды микроскопии и такие методы, как оже-спектрометрия, фотоакустическая спектроскопия, компьютерная томография, измерения радиоактивности и др.
Эффективным недеструктивным методом является отражательная инфракрасная спектроскопия, которая используется для определения примесей различных продуктов разложения и воды, а также в анализе многокомпонентных смесей.
4.3 Абсорбционные методыАбсорбционные методы основаны на свойствах веществ поглощать свет в различных областях спектра.
Атомно-абсорбционная спектрофотометрия основана на использовании ультрафиолетового или видимого излучения резонансной частоты. Поглощение излучения вызывается переходом электронов с внешних орбиталей атомов на орбитали с более высокой энергией. Объектами, поглощающими излучение, являются газообразные атомы, а также некоторые органические вещества. Сущность определений методом атомно-абсорбционной спектрометрии состоит в том, что через пламя, в котором распыляется анализируемый раствор пробы, проходит резонансное излучение от лампы с полым катодом. Это излучение попадает на входную щель монохроматора, причем из спектра выделяется только резонансная линия испытуемого элемента. Фотоэлектрическим методом измеряют уменьшение интенсивности резонансной линии, происходящей вследствие поглощения ее атомами определяемого элемента. Расчет концентрации производят с помощью уравнения, отражающего ее зависимость от ослабления интенсивности излучения источника света, длины поглощающего слоя и коэффициента поглощения света в центре линии поглощения. Метод отличается высокой избирательностью и чувствительностью.
Поглощение резонансных линий измеряют на атомно-абсорбцион- ных спектрофотометрах типа "Спектр-1", "Сатурн" и др. Точность определений не превышает 4%, предел обнаружения достигает 0,001 мкг/мл. Это свидетельствует о высокой чувствительности метода. Он находит все более широкое применение для оценки чистоты лекарственных препаратов, в частности определения минимальных примесей тяжелых металлов. Перспективно использование атомно-абсорбционной спектрофотометрии для анализа поливитаминных препаратов, аминокислот, барбитуратов, некоторых антибиотиков, алкалоидов, галогенсодержащих лекарственных веществ, ртутьсодержащих соединений.
Возможно также применение в фармации рентгеновской абсорбционной спектроскопии, основанной на поглощении атомами рентгеновского излучения.
Ультрафиолетовая спектрофотометрия — наиболее простой и широко применяемый в фармации абсорбционный метод анализа. Его используют на всех этапах фармацевтического анализа лекарственных препаратов (испытания подлинности, чистоты, количественное определение). Разработано большое число способов качественного и количественного анализа лекарственных форм методом ультрафиолетовой спектрофотометрии. Для идентификации могут быть использованы атласы спектров лекарственных веществ, систематизирующие сведения о характере спектральных кривых и значениях удельных показателей поглощения.
Известны различные варианты использования метода УФ-спектрофотометрии для идентификации. При испытаниях на подлинность идентифицируют лекарственные вещества по положению максимума светопоглощения. Чаще в фармакопейных статьях приведены положения максимума (или минимума) и соответствующие им значения оптических плотностей. Иногда используют метод, основанный на вычислении отношения оптических плотностей при двух длинах волн (они обычно соответствуют двум максимумам или максимуму и минимуму светопоглощения). Идентифицируют целый ряд лекарственных веществ также по удельному показателю поглощения раствора.
Весьма перспективно для идентификации лекарственных веществ использование таких оптических характеристик, как положение полосы поглощения в шкале длин волн, частота в максимуме поглощения, значение пиковой и интегральной интенсивности, полуширина и асимметрия полос, сила осциллятора. Эти параметры делают более надежной идентификацию веществ, чем установление длины волны максимума светопоглощения и удельного показателя поглощения. Эти константы, позволяющие охарактеризовать наличие связи между УФ-спектром и структурой молекулы, были установлены и использованы для оценки качества лекарственных веществ, содержащих гетероатом кислорода в молекуле (В.П.Буряк).
Объективный выбор оптимальных условий количественного спектрофотометрического анализа можно осуществить только предварительным исследованием констант ионизации, влияния природы растворителей, рН среды и других факторов на характер спектра поглощения.
В НТД приведены различные способы использования УФ-спектрофотометрии для количественного определения лекарственных веществ, являющихся витаминами (ретинола ацетат, рутин, цианокобаламин), стероидными гормонами (кортизона ацетат, преднизон, прегнин, тестостерона пропионат), антибиотиками (натриевые соли оксациллина и метициллина, феноксиметилпенциллин, левомицетина стеарат, гризеофульвин). В качестве растворителей для спектрофотометрических измерений обычно используют воду или этанол. Расчет концентрации проводят различными способами: по стандарту, удельному показателю поглощения или калибровочному графику.
Количественный спектрофотометрический анализ целесообразно комбинировать с установлением подлинности по УФ-спектру. В этом случае раствор, приготовленный из одной навески, можно использовать для обоих этих испытаний. Чаще всего при спектрофотометрических определениях применяют способ, основанный на сравнении оптических плотностей анализируемого и стандартного растворов. Определенных условий анализа требуют лекарственные вещества, способные образовывать кислотно-основные формы в зависимости от рН среды. В таких случаях необходимо предварительно подбирать условия, в которых вещество в растворе полностью будет находиться в одной из таких форм.
Для уменьшения относительной погрешности фотометрического анализа, в частности снижения систематической ошибки, весьма перспективно использование стандартных образцов лекарственных веществ. Учитывая сложность получения и высокую стоимость, они могут быть заменены эталонами, приготавливаемыми из доступных неорганических соединений (дихромата калия, хромата калия).
В ГФ XI расширена область применения УФ-спектрофотометрии. Метод рекомендован для анализа многокомпонентных систем, а также для анализа лекарственных веществ, которые сами не поглощают свет в ультрафиолетовой и видимой областях спектра, но могут быть превращены в поглощающие свет соединения с помощью различных химических реакций.
Дифференциальные методы позволяют расширить область применения фотометрии в фармацевтическом анализе. Они дают возможность повысить ее объективность и точность, а также анализировать высокие концентрации веществ. Кроме того, этими методами можно анализировать многокомпонентные смеси без предварительного разделения.
Метод дифференциальной спектрофотометрии и фотоколориметрии включен в ГФ XI, вып. 1 (с. 40). Сущность его заключается в измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество испытуемого вещества. Это приводит к изменению рабочей области шкалы прибора и снижению относительной погрешности анализа до 0,5—1%, т.е. такой же, как и у титриметрических методов. Хорошие результаты были получены при использовании вместо растворов сравнения нейтральных светофильтров с известной оптической плотностью; входящих в комплект спектрофотометров и фотоколориметров (В.Г.Беликов).
Дифференциальный метод нашел применение не только в спектрофотометрии и фотоколориметрии, но и в фототурбидиметрии, фотонефелометрии, интерферометрии. Дифференциальные методы могут быть распространены и на другие физико-химические методы. Большие перспективы для анализа лекарств имеют и методы химического дифференциального анализа, основанные на использовании таких химических воздействий на состояние лекарственного вещества в растворе, как изменение рН среды, смена растворителя, изменение температуры, влияние электрических, магнитных, ультразвуковых полей и др.
Широкие возможности открывает в количественном спектрофотометрическом анализе один из вариантов дифференциальной спектрофотометрии — ΔЕ-метод. Он основан на превращении анализируемого вещества в таутомерную (или иную) форму, отличающуюся по характеру светопоглощения.
Новые возможности в области идентификации и количественного определения органических веществ открывает использование производной УФ-спектрофотометрии. Метод основан на выделении индивидуальных полос из УФ-спектров, представляющих собой сумму налагающихся полос поглощения или полос, не имеющих четко выраженного максимума поглощения.
Производная спектрофотометрия дает возможность идентификации сходных по химической структуре лекарственных веществ или их смесей. Для повышения избирательности качественного спектрофотометрического анализа применяют способ построения вторых производных УФ-спектров. Вторую производную можно рассчитать способом численного дифференцирования.
Разработан унифицированный метод получения производных от спектров поглощения, который учитывает особенности характера спектра. Показано, что вторая производная имеет разрешающую способность примерно в 1,3 раза больше по сравнению с непосредственной спектрофотометрией. Это позволило использовать данный метод для идентификации кофеина, теобромина, теофиллина, папаверина гидрохлорида и дибазола в лекарственных формах. Вторая и четвертая производные в количественном анализе более эффективны по сравнению с титриметрическими методами. Продолжительность определения сокращается в 3-4 раза. Определение указанных препаратов в смесях оказалось возможным вне зависимости от характера поглощения сопутствующих веществ или при существенном уменьшении влияния их светопоглощения. Это позволяет исключить трудоемкие операции по разделению смесей.
Использование в спектрофотометрическом анализе комбинированного полинома позволило исключить влияние нелинейного фона и разработать методики количественного определения ряда препаратов в лекарственных формах, не требующие сложных расчетов результатов анализа. Комбинированный полином успешно применен при изучении процессов, происходящих при хранении лекарственных веществ и в химико-токсикологических исследованиях, так как позволяет уменьшить влияние светопоглощающих примесей (Е.Н.Вергейчик).
Спектроскопия комбинационного рассеяния (СКР) отличается от других спектроскопических методов по чувствительности, большому выбору растворителей и интервалов температур. Наличие отечественного КР-спектрометра марки ДСФ-24 позволяет применять этот метод не только для установления химической структуры, но и в фармацевтическом анализе.
Не получил еще должного развития в практике фармацевтического анализа метод спектрофотометрического титрования. Этот метод дает возможность выполнения безындикаторного титрования многокомпонентных смесей с близкими значениями рК на основе последовательного изменения оптической плотности в процессе титрования в зависимости от объема добавляемого титранта.
Фотоколориметрический метод широко применяется в фармацевтическом анализе. Количественное определение этим методом в отличие от УФ-спбктрофотометрии осуществляют в видимой области спектра. Определяемое вещество с помощью какого-либо реагента переводят в окрашенное соединение, а затем измеряют интенсивность окраски раствора на фотоколориметре. Точность определений зависит от выбора оптимальных условий протекания химической реакции.
Очень широко в фотометрическом анализе используются методики анализа препаратов, производных первичных ароматических аминов, основанные на использовании реакций диазотирования и азосочетания. В качестве азосоставляющего широко применяют N-(1-нафтил)-этилендиамин. Реакция образования азокрасителей лежит в основе фотометрического определения многих препаратов, производных фенолов.
Фотоколориметрический метод включен в НТД для количественного определения ряда нитропроизводных (нитроглицерин, фурадонин, фуразолидон), а также препаратов витаминов (рибофлавин,фолиевая кислота) и сердечных гликозидов (целанид). Разработаны многочисленные методики фотоколориметрического определения препаратов в лекарственных формах. Известны различные модификации фотоколориметрии и способы расчета концентрации в фотоколориметрическом анализе.
Перспективными для применения в качестве цветореагентов в фотометрическом анализе оказались такие поликарбонильные соединения, как биндон (ангидро-бис-индандион-1,3), аллоксан (тетраоксогекса-гидропиримидин), натриевая соль 2-карбэтоксииндандиона-1,3 и некоторые ее производные. Установлены оптимальные условия и разработаны унифицированные способы идентификации и спектрофотометрического определения в видимой области лекарственных веществ, содержащих первичную ароматическую или алифатическую аминогруппу, остаток сульфонил мочевины или являющимися азотсодержащими органическими основаниями и их солями (В.В.Петренко).
Широко используют в фотоколориметрии реакции окрашивания, основанные на образовании полиметиновых красителей, которые получаются при разрыве пиридинового или фуранового циклов либо при некоторых реакциях конденсации с первичными ароматическими аминами (А.С.Бейсенбеков).
Для идентификации и спектрофотометрического определения в видимой области спектра лекарственных веществ, производных ароматических аминов, тиолов, тиоамидов и других меркаптосоединений использованы в качестве цветореагентов N-хлор-, N-бензолсульфонил- и N-бензолсульфонил-2-хлор-1,4-бензохинонимина.
Один из вариантов унификации способов фотометрического анализа основан на косвенном определении по остатку нитрита натрия, вводимого в реакционную смесь в виде стандартного раствора, взятого в избытке. Избыток нитрита определяют затем фотометрически реакцией диазотирования с помощью этакридина лактата. Такой прием применен для косвенного фотометрического определения азотсодержащих лекарственных веществ по нитрит-иону, образующемуся в результате их превращений (гидролиза, термического разложения). Унифицированная методика позволяет осуществлять контроль качества более 30 таких лекарственных веществ в многочисленных лекарственных формах (П.Н.Ивахненко).
Фототурбидиметрия и фотонефелометрия – это методы, имеющие большие возможности, но пока ограниченно применяющиеся в фармацевтическом анализе. Основаны на измерении света, поглощенного (турбидиметрия) или рассеянного (нефелометрия) взвешенными частицами анализируемого вещества. С каждым годом методы совершенствуются. Рекомендуют, например, хронофототурбидиметрию в анализе лекарственных веществ. Сущность метода заключается в установлении изменений светопогашений во времени. Описано также применение термонефелометрии, основанной на установлении зависимости концентрации вещества от температуры, при которой наступает помутнение раствора препарата.
Систематические исследования в области фототурбидиметрии, хронофототурбидиметрии и фототурбидиметрического титрования показали возможность применения фосфорно-вольфрамовой кислоты для количественного определения азотсодержащих лекарственных веществ. В фототурбидиметрическом анализе использован как непосредственный, так и дифференциальный метод, а также автоматическое фототурбидиметрическое титрование и хронофототурбидиметрическое определение двухкомпонентных лекарственных форм (А.И.Сичко).
Инфракрасная (ИК) спектроскопия характеризуется широкой информативностью, что создает возможность объективной оценки подлинности и количественного определения лекарственных веществ. ИК-спектр однозначно характеризует всю структуру молекулы. Различия в химическом строении меняют характер ИК-спектра. Важные преимущества ИК-спектрофотометрии — специфичность, быстрота выполнения анализа, высокая чувствительность, объективность получаемых результатов, возможность анализа вещества в кристаллическом состоянии.
ИК-спектры измеряют, используя обычно взвеси лекарственных веществ в вазелиновом масле, собственное поглощение которого не мешает идентификации анализируемого соединения. Для установления подлинности используют, как правило, расположенную в интервале частот от 650 до 1800 см-1 так называемую область "отпечатков пальцев" (650—1500 см-1), а также валентные колебания химических связей
С=0, С=С, С=N
В ГФ XI рекомендованы два способа установления подлинности лекарственных веществ но ИК-спектрам. Один из них основан на сравнении ИК-спектров испытуемого вещества и его стандартного образца. Спектры должны быть сняты в идентичных условиях, т.е. образцы должны быть в одинаковом агрегатном состоянии, в одной и той же концентрации, единой должна быть скорость регистрации и т.д. Второй способ заключается в сравнении ИК-спектра испытуемого вещества с его стандартным спектром. В этом случае необходимо строго соблюдать условия, предусмотренные для снятия стандартного спектра, приведенные в соответствующей НТД (ГФ, ВФС, ФС). Полное совпадение полос поглощения свидетельствует об идентичности веществ. Однако полиморфные модификации могут давать различные ИК-спектры. В таком случае для подтверждения идентичности необходимо перекристаллизовать испытуемые вещества из одного и того же растворителя и вновь снять спектры.
Подтверждением подлинности лекарственного вещества может служить также интенсивность поглощения. Для этой цели используют такие константы как показатель поглощения или величина интегральной интенсивности поглощения, равная площади, которую огибает кривая на спектре поглощения.
Установлена возможность использования ИК-спектроскопии для идентификации большой группы лекарственных веществ, содержащих в молекуле карбонильные группы. Подлинность устанавливают по характеристическим полосам поглощения в следующих областях: 1720-1760, 1424-1418, 950-в00 см-1 для карбоновых кислот; 1596-1582, 1430-1400, 1630-1612, 1528-1518 см-1 для аминокислот; 1690—1670, 1615—1580 см-1 для амидов; 1770—1670 см-1 для производных барбитуровой кислоты; 1384—1370, 1742—1740, 1050 см-1 для терпеноидов; 1680—1540, 1380—1278 см-1 для антибиотиков тетрациклинового ряда; 3580-3100, 3050-2870, 1742-1630, 903-390 см-1 для стероидов (А.Ф.Мынка).
Метод ИК-спектроскопии включен в фармакопеи многих зарубежных стран и в МФ III, где использован для идентификации более 40 лекарственных веществ. Методом ИК-спектрофотометрии можно проводить не только количественную оценку лекарственных веществ, но и исследование таких химических превращений, как диссоциация, сольволиз, метаболизм, полиморфизм и т.д.
4.4 Методы, основанные на испускании излученияК этой группе методов относят фотометрию пламени, флуоресцентные и радиохимические методы.
В ГФ XI включена эмиссионная и пламенная спектрометрия для целей качественного и количественного определения химических элементов и их примесей в лекарственных веществах. Измерение интенсивности излучения спектральных линий испытуемых элементов выполняют на отечественных пламенных фотометрах ПФЛ-1, ПФМ, ПАЖ-1. Регистрирующими системами служат фотоэлементы, связанные с цифровыми и печатающими устройствами. Точность определений методами эмиссионной, как и атомно-абсорбционной, пламенной спектрометрии находится в пределах 1—4%, предел обнаружения может достигать 0,001 мкг/мл.
Количественное определение элементов методом эмиссионной пламенной спектрометрии (пламенной фотометрии) основано на установлении зависимости между интенсивностью спектральной линии и концентрацией элемента в растворе. Сущность выполнения испытания состоит в распылении анализируемого раствора до состояния аэрозоля в пламени горелки. Под воздействием температуры пламени происходят испарение растворителя и твердых частиц из капель аэрозоля, диссоциация молекул, возбуждение атомов и возникновение их характеристического излучения. С помощью светофильтра или монохроматора излучение анализируемого элемента отделяется от других и, попадая на фотоэлемент, вызывает фототок, который измеряется с помощью гальванометра или потенциометра.
Пламенная фотометрия использована для количественного анализа натрий-, калий- и кальций-содержащих препаратов в лекарственных формах. На основе исследования влияния на эмиссию определяемых катионов, органических анионов, вспомогательных и сопутствующих компонентов были разработаны методики количественного определения натрия гидрокарбоната, натрия салицилата, ПАСК-натрия, билигноста, гексенала, натрия нуклеината, кальция хлорида и глюконата, бепаска и др. Предложены методики одновременного определения двух солей с разными катионами в лекарственных формах, например калия иодида — натрия гидрокарбоната, кальция хлорида — калия бромида, калия иодида — натрия салицилата и др.
Люминесцентные методы основаны на измерении вторичного излучения, возникающего в результате воздействия света на анализируемое вещество. К их числу относят флуоресцентные методы, хемилюминесценцию, рентгенофлуоресценцию и др.
Флуоресцентные методы основаны на способности веществ флуоресцировать в УФ-свете. Эта способность обусловлена структурой либо самих органических соединений, либо продуктов их диссоциации, сольволиза и других превращений, вызванных воздействием различных реактивов.
Флуоресцирующими свойствами обладают обычно органические соединения с симметричной структурой молекул, в которых имеются сопряженные связи, нитро-, нитрозо-, азо-, амидо-, карбоксильная или карбонильная группы. Интенсивность флуоресценции зависит от химической структуры и концентрации вещества, а также других факторов.
Флуориметрия может быть использована как для качественного, так и для количественного анализа. Количественный анализ выполняют на спектрофлуориметрах. Принцип их работы состоит в том, что свет от ртутно-кварцевой лампы через первичный светофильтр и конденсор падает на кювету с раствором испытуемого вещества. Расчет концентрации проводят по шкале стандартных образцов флуоресцирующего вещества известной концентрации.
Разработаны унифицированные методики количественного спект- рофлуориметрического определения производных п-аминобензолсульфамида (стрептоцид, сульфацил-натрий, сульгин, уросульфан и др.) и п-аминобензойной кислоты (анестезин, новокаин, новокаинамид). Водно-щелочные растворы сульфаниламидов имеют наибольшую флуоресценцию при рН б—8 и 10—12. Кроме того, сульфаниламиды, содержащие в молекуле незамещенную первичную ароматическую аминогруппу, после нагревания с о-фталевым альдегидом в присутствии серной кислоты приобретают интенсивную флуоресценцию в области 320—540 нм. В той же области флуоресцируют производные барбитуровой кислоты (барбитал, барбитал-натрий, фенобарбитал, этаминал-натрий) в щелочной среде (рН 12—13) с максимумом флуоресценции при 400 нм. Предложены высокочувствительные и специфичные методики спектрофлуориметрического определения антибиотиков: тетрациклина, окситетрациклина гидрохлорида, стрептомицина сульфата, пассомицина, флоримицина сульфата, гризеофульвина и сердечного гликозида целанида (Ф.В.Бабилев). Проведены исследования спектров флуоресценции ряда лекарственных средств, содержащих природные соединения: производные кумарина, антрахинона, флавоноидов (В.П.Георгиевский).
Выявлены комплексообразующие группировки у 120 лекарственных веществ, производных оксибензойной, оксинафтойной, антраниловой кислот, 8-оксихинолина, оксипиридина, 3- и 5-оксифлавона, птеридина и др. Указанные группировки способны образовывать флуоресцирующие комплексы с катионами магния, алюминия, бора, цинка, скандия при возбуждении флуоресценции от 330 нм и выше и ее излучении при длинах волн, превышающих 400 нм. Проведенные исследования позволили разработать методики флуориметрирования 85 лекарственных средств (А.А.Хабаров).
Наряду с производной спектрофотометрией в фармацевтическом анализе обоснована возможность применения производной спектрофлуориметрии. Спектры снимают на флуоресцентном спектрофотометре МПФ-4 с термостатирующей ячейкой, а производные находят аналогичным дифференцированием с помощью компьютера. Метод использован для разработки простых, точных и высокочувствительных методик количественного определения гидрохлоридов пиридоксина и эфедрина в лекарственных формах в присутствии продуктов разложения.
Перспективность использования рентгеновской флуоресценции для определения малых количеств примесей в лекарственных препаратах обусловливается высокой чувствительностью и возможностью выполнения анализа без предварительного разрушения вещества. Метод рентгенофлуоресцентной спектрометрии оказался перспективным для количественного анализа веществ, имеющих в молекуле такие гетероатомы, как железо, кобальт, бром, серебро и др. Принцип метода заключается в сравнении вторичного рентгеновского излучения элемента в анализируемом и стандартном образце. Рентгенофлуоресцентная спектрометрия относится к числу методов, не требующих предварительных деструктивных изменений. Выполняют анализ на отечественном спектрометре РС-5700. Продолжительность анализа 15 мин.
Хемилюминесценция — метод, заключающийся в использовании энергии, возникающей в процессе химических реакций.
Эта энергия служит источником возбуждения. Ее излучают при окислении некоторые барбитураты (особенно фенобарбитал), гидразиды ароматических кислот и другие соединения. Это создает большие возможности использования метода для определения очень малых концентраций веществ в биологическом материале.
Радиохимические методы находят все более широкоеприменение в фармацевтическоманализе. Радиометрический анализ, основанный на измерении β- или γ-излучения с помощью спектрометров, использован (наряду с другими параметрами для оценки качества фармакопейных радиоактивных препаратов. Широко применяют в различных областях техники и особенно в аналитической химии высокочувствительные методы анализа с применением радиоактивных изотопов (меченых атомов). Для обнаружения следов примесей в веществах используют активационный анализ; для определения в смесях близких по свойствам трудноразделяемых компонентов — метод изотопного разбавления. Применяют также радиометрическое титрование и радиоактивные индикаторы. Оригинальным вариантом сочетания радиоизотопного и хроматографического методов является изучение диффузионно-осадочных хроматограмм в тонком слое желатинового геля с помощью радиоактивных индикаторов.
4.5 Методы, основанные на использовании магнитного поляМетоды ЯМР-, ПМР-спектроскопии, а также масс-спектрометрии отличаются высокой специфичностью, чувствительностью и используются для анализа многокомпонентных смесей, в том числе лекарственных форм без предварительного их разделения.
Метод спектроскопии ЯМР используют для испытания подлинности лекарственных веществ, которая может быть подтверждена либо по полному набору спектральных параметров, характеризующих структуру данного соединения, либо по наиболее характерным сигналам спектра. Подлинность можно также установить с помощью стандартного образца, добавляя определенное его количество к анализируемому раствору. Полное совпадение спектров анализируемого вещества и его смеси со стандартным образцом указывает на их идентичность.
Регистрацию ЯМР-спектров выполняют на спектрометрах с рабочими частотами 60 мГц и более, используя такие основные характеристики спектров, как химический сдвиг, мультиплетность сигнала резонанса, константу спин-спинового взаимодействия, площадь сигнала резонанса. Наиболее обширную информацию о молекулярной структуре анализируемого вещества дают спектры ЯМР 13С и 1Н.
Надежная идентификация препаратов гестагенных и эстрогенных гормонов, а также их синтетических аналогов: прогестерона, прегнина, этинилэстрадиола, метилэстрадиола, эстрадиола дипропионата и др. — может быть осуществлена методом спектроскопии ЯМР 1Н в деитерированном хлороформе на спектрометре УН-90 с рабочей частотой 90 мГц (внутренний стандарт — тетраметилсилан).
Систематические исследования позволили установить возможность применения спектроскопии ЯМР 13С для идентификации лекарственных веществ 10-ацилпроизводных фенотиазина (хлорацизина, фторацизина, этмозина, этацизина), 1,4-бензодиазепина (хлор-, бром- и нитропроизводные) и др. Методом спектроскопии ЯМР 1Н и 13С осуществлены идентификация, количественная оценка основных компонентов и примесей в препаратах и стандартных образцах природных и полусинтетических антибиотиков аминогликозидов, пенициллинов, цефалоспоринов, макролидов и др. Указанный метод использован для идентификации в унифицированных условиях ряда витаминов: липоевой и аскорбиновой кислот, липамида, холина и метилметионинсульфония хлоридов, ретинола пальмитата, кальция пантотената, эргокальциферола. Метод спектроскопии ЯМР 1Н позволил осуществлять надежную идентификацию таких сложных по химической структуре природных соединений, как сердечные гликозиды (дигоксин, дигитоксин, целанид, дезланозид, нериолин, цимарин и др.). Для ускорения обработки спектральной информации использована ЭВМ. Ряд методик идентификации включен в ФС и ВФС (В.С.Карташов).
Количественное определение лекарственного вещества может быть также выполнено с использованием спектров ЯМР. Относительная погрешность количественных определений методом ЯМР зависит от точности измерений площадей резонансных сигналов и составляет ±2—5%. При определении относительного содержания вещества или его примеси измеряют площади сигналов резонанса испытуемого вещества и стандартного образца. Затем вычисляют количество испытуемого вещества. Для определения абсолютного содержания лекарственного вещества или примеси анализируемые образцы готовят количественно и добавляют к навеске точно отвешенную массу внутреннего стандарта. После этого выполняют регистрацию спектра, измеряют площади сигналов анализируемого вещества (примеси) и внутреннего стандарта, затем вычисляют абсолютное содержание.
Развитие импульсной техники Фурье-спектроскопии, применение ЭВМ позволили резко повысить чувствительность метода ЯМР 13С и распространить его на количественный анализ многокомпонентных смесей биоорганических соединений, в том числе лекарственных веществ без их предварительного разделения.
Спектроскопические параметры ПМР-спектров дают целый комплекс разнообразной и весьма селективной информации, который может быть использован в фармацевтическом анализе. Следует строго соблюдать условия регистрации спектров, так как на значения химических сдвигов и другие параметры оказывают влияние тип растворителя, температура, рН раствора, концентрация вещества.
Если полная интерпретация ПМР-спектров затруднена, то выделяют только характерные сигналы, по которым идентифицируют испытуемое вещество. ПМР-спектроскопия применена для испытания подлинности многих лекарственных веществ, в том числе барбитуратов, гормональных средств, антибиотиков и др.
Поскольку метод дает информацию о наличии или отсутствии примесей к основному веществу, важное практическое значение имеет ПМР-спектроскопия для испытания лекарственных веществ на чистоту. Различия в значениях величин тех или иных констант позволяют сделать заключение о присутствии примесей продуктов разложения лекарственного вещества. Чувствительность метода к примесям колеблется в широких пределах и зависит от спектра основного вещества, наличия в молекулах тех или иных групп, содержащих протоны, растворимости в соответствующих растворителях. Минимальное содержание примеси, которое можно установить, составляет обычно 1—2%. Особенно ценной является возможность обнаружения примесей изомеров, присутствие которых невозможно подтвердить другими методами. Так, например, обнаружена примесь кислоты салициловой в кислоте ацетилсалициловой, морфина в кодеине и т.д.
Количественный анализ на основе использования ПМР-спектроскопии имеет преимущества перед другими методами, заключающиеся в том, что при анализе многокомпонентных смесей нет необходимости выделять индивидуальные компоненты для калибровки прибора. Поэтому метод широко применим для количественного анализа как индивидуальных лекарственных веществ, так и растворов, таблеток, капсул, суспензий и других лекарственных форм, содержащих один или несколько ингредиентов. Стандартное отклонение не превышает ±2,76%. Описаны способы анализа таблеток фуросемида, мепробамата, хинидина, преднизолона и др.
Расширяется диапазон применения масс-спектрометрии в анализе лекарственных веществ для идентификации и количественного анализа. Метод основан на ионизации молекул органических соединений. Он отличается большой информативностью и исключительно высокой чувствительностью. Масс-спектрометрию применяют для определения антибиотиков, витаминов, пуриновых оснований, стероидов, аминокислот и других лекарственных веществ, а также продуктов их метаболизма.
Использование лазеров в аналитических приборах значительно расширяет практическое применение УФ- и ИК-спектрофотометрии, а также флуоресцентной и масс-спектроскопии, спектроскопии комбинационного рассеяния, нефелометрии и других методов. Лазерные источники возбуждения позволяют повысить чувствительность многих методов анализа, сократить продолжительность их выполнения. Лазеры используют в дистанционном анализе в качестве детекторов в хроматографии, в биоаналитической химии и т.д.
4.6 Электрохимические методыЭта группа методов качественного и количественного анализа основана на электрохимических явлениях, происходящих в исследуемой среде и связанных с изменениями химической структуры, физических свойств или концентрации веществ.
Потенциометрия — метод, основанный на измерении равновесных потенциалов, возникающих на границе между испытуемым раствором и погруженным в него электродом. В ГФ XI включен метод потенциометрического титрования, заключающийся в установлении эквивалентного объема титранта путем измерения ЭДС индикаторного электрода и электрода сравнения, погруженных в анализируемый раствор. Метод прямой потенциометрии используется для определения рН (рН-метрия) и установления концентрации отдельных ионов. Потенциометрическое титрование отличается от индикаторного возможностью анализировать сильно окрашенные, коллоидные и мутные растворы, а также растворы, содержащие окислителй. Кроме того, можно последовательно оттитровать в смеси несколько компонентов в водных и неводных средах. Потенциометрический метод используют для титрования на основе реакций нейтрализации, осаждения, комплексообразования, окисления — восстановления. Электродом сравнения во всех указанных методах служит каломельный, хлорсеребряный или стеклянный (последний не используют при анализе методом нейтрализации). Индикаторным при кислотно-основном титровании является стеклянный электрод, при комплексонометрическом — ртутный или ион-селективный, в методе осаждения — серебряный, в окислительно-восстановительном — платиновый.
Измерение ЭДС, возникающей при титровании за счет разности потенциалов между индикаторным электродом и электродом сравнения, производят с помощью высокоомных рН-метров. Титрант прибавляют из бюретки равными объемами, постоянно перемешивая титруемую жидкость. Вблизи точки эквивалентности титрант прибавляют по 0,1—0,05 мл. Значение ЭДС в этой точке изменяется наиболее сильно, так как абсолютная величина отношения изменения ЭДС к приращению объема прибавляемого титранта будет при этом максимальной. Результаты титрования представляют либо графически, устанавливая точку эквивалентности на кривой титрования, либо расчетным методом. Затем вычисляют эквивалентный объем титранта по формулам (см. ГФ XI, вып. 1, с. 121).
Амперометрическое титрование с двумя индикаторными электродами, или титрование "до полного прекращения тока", основано на использовании пары идентичных инертных электродов (платина, золото), которые находятся под небольшим напряжением. Метод наиболее часто используют для нитрито- и иодометрического титрования. Точку эквивалентности находят по резкому увеличению силы тока, проходящего через ячейку (в течение 30 с) после добавления последней порции реагента. Эту точку можно установить графическим методом по зависимости силы тока от объема добавленного реагента, так же как при потенциометрическом титровании (ГФ XI, вып. 1, с. 123). Разработаны также способы биамперометрического титрования лекарственных веществ при использовании методов нитритометрии, осаждения и окисления — восстановления.
Особенно перспективна ионометрия, использующая зависимость между ЭДС гальванической сети с ионоселективным электродом и концентрацией анализируемого иона в электродной ячейке цепи. Определения неорганических и органических (азотсодержащих) лекарственных веществ с помощью ионоселективных электродов отличаются от других методов высокой, чувствительностью, экспрессностью, хорошей воспроизводимостью результатов, несложным оборудованием, доступными реагентами, пригодностью для автоматизированного контроля и исследования механизма действия лекарств. В качестве примера можно привести способы ионометрического определения калия, натрия, галогенидов и кальцийсодержащих лекарственных веществ в таблетках и в солевых кровезамещающих жидкостях. С помощью отечественных рН-метров (рН-121, рН-673), ионометра И-115 и калий селективных электродов определяют калиевые соли различных кислот (оротовой, аспарагиновой и др.).
Полярография — метод анализа, основанный на измерении силы тока, возникающего на микроэлектроде при электровосстановлении или электроокислении анализируемого вещества в растворе. Электролиз проводят в полярографической ячейке, которая состоит из электролизера (сосуда) и двух электродов. Один из них — ртутный капающий микроэлектрод, а другой — макроэлектрод, которым служит либо слой ртути на электролизере, либо внешний насыщенный каломельный электрод. Полярографический анализ может быть выполнен в водной среде, в смешанных растворителях (вода — этанол, вода — ацетон), в неводных средах (этаноле, ацетоне, диметилформамиде и др.). При идентичных условиях измерений для идентификации вещества используют потенциал полуволны. Количественное определение основано на измерении предельного диффузного тока испытуемого лекарственного вещества (высота волны). Для определения содержания используют метод калибровочных кривых, метод стандартных растворов и метод добавок (ГФ XI, вып. 1, с. 154). Полярографию широко используют в анализе неорганических веществ, а также алкалоидов, витаминов, гормонов, антибиотиков, сердечных гликозидов. Весьма перспективны вследствие высокой чувствительности современные методы: дифференциальная пульс-полярография, осциллографическая полярография и др.
Далеко не исчерпаны возможности электрохимических методов в фармацевтическом анализе. Разрабатываются новые варианты потенциометрии: инверсионная бестоковая хронопотенциометрия, прямая потенциометрия с помощью газового аммоний-селективного электрода и др. Расширяются исследования в области применения в фармацевтическом анализе таких методов, как кондуктометрия, основанная на исследовании электрической проводимости растворов анализируемых веществ; кулонометрия, заключающаяся в измерении количества электричества, затраченного на электрохимическое восстановление или окисление определяемых ионов.
Кулонометрия имеет ряд преимуществ перед другими физико-химическими и химическими методами. Поскольку этот метод основан на измерении количества электричества, он дает возможность непосредственно определять массу вещества, а не какое-либо свойство, пропорциональное концентрации. Вот почему кулонометрия исключает необходимость использования не только стандартных, но и титрованных растворов. Что касается кулонометрического титрования, то оно расширяет область титриметрии за счет применения различных неустойчивых электрогенерированных титрантов. Одна и та же электрохимическая ячейка может быть использована для проведения титрования с использованием различных типов химических реакций. Так, методом нейтрализации можно опредачить кислоты и основания даже в миллимолярных растворах с погрешностью не более 0,5%.
Кулонометрический метод применяют при определении малых количеств анаболических стероидов, местно-анестезирующих и других лекарственных веществ. Определению не мешают наполнители таблеток. Методики отличаются простотой, экспрессностью, быстротой и чувствительностью.
Метод диэлектрических измерений в диапазоне электромагнитных волн широко применяют для экспресс-анализа в химической технологии, пищевой промышленности и других областях. Одним из перспективных направлений является диэлькометрический контроль ферментных и других биопрепаратов. Он позволяет осуществить быструю, точную, безреагентную оценку таких параметров, как влажность, степень гомогенности и чистоты препарата. Диэлькометрический контроль является многопараметровым, испытуемые растворы могут быть непрозрачными, а измерения можно выполнять бесконтактным способом с записью результатов на ЭВМ.
4.7 Методы разделенияИз физико-химических методов разделения в фармацевтическом анализе в основном используют хроматографию, электрофорез и экстракцию.
Хроматографические методы разделения веществ основаны на их распределении между двумя фазами: подвижной и неподвижной. Подвижной фазой может быть жидкость или газ, неподвижной — твердое вещество или жидкость, адсорбированная на твердом носителе. Относительная скорость перемещения частиц вдоль пути разделения зависит от взаимодействия их с неподвижной фазой. Это приводит к тому, что каждое из веществ проходит определенную длину пути на носителе. Отношение скорости перемещения вещества к скорости перемещения растворителя обозначают Эта величина является константой вещества для данных условий разделения и используется для идентификации.
Хроматография дает возможность наиболее эффективно осуществлять избирательное распределение компонентов анализируемого образца. Это имеет существенное значение для фармацевтического анализа, объектами исследования в котором обычно являются смеси нескольких веществ.
По механизму процесса разделения хроматографические методы классифицируют на ионообменную, адсорбционную, осадочную, распределительную, окислительно-восстановительную хроматографию. По форме проведения процесса можно выделить колоночную, капиллярную и плоскостную хроматографию. Последняя может быть выполнена на бумаге и в тонком (закрепленном или незакрепленном) слое сорбента. Хроматографические методы классифицируют также по агрегатно- му состоянию анализируемого вещества. К ним относятся различные методы газовой и жидкостной хроматографии.
Адсорбционная хроматография основана на избирательной адсорбции отдельных компонентов из раствора смеси веществ. Стационарной фазой служат такие адсорбенты, как оксид алюминия, активированный уголь и др.
Ионообменная хроматография использует ионообменные процессы, происходящие между адсорбентом и ионами электролита в анализируемом растворе. Стационарной фазой служат катион обменные или ани- онобменные смолы, содержащиеся в них ионы способны обмениваться на одноименно заряженные противоионы.
Осадочная хроматография основана на различии в растворимости веществ, образующихся при взаимодействии компонентов разделяемой смеси с осадителем.
Распределительная хроматография заключается в распределении компонентов смеси между двумя несмешивающимися жидкими фазами (подвижной и неподвижной). Стационарной фазой служит пропитанный растворителем носитель, а подвижной фазой — органический растворитель, практически не смешивающийся с первым растворителем. При выполнении процесса в колонке происходит разделение смеси на зоны, содержащие по одному компоненту. Распределительная хроматография может выполняться также в тонком слое сорбента (тонкослойная хроматография) и на хроматографической бумаге (бумажная хроматография).
Ранее других методов разделения в фармацевтическом анализе йачали применять ионообменную хроматографию для количественного определения препаратов: солей серной, лимонной и других кислот. При этом ионообменную хроматографию сочетают с кислотно-основным титрованием. Совершенствование метода позволило, используя хроматографию ионных пар с обращенной фазой, разделять некоторые гидрофильные органические соединения. Возможно сочетание комплексонометрии с использованием катионитов в Zn2+-фopмe для анализа аминопроизводных в смесях и алкалоидов в экстрактах и настойках. Таким образом, сочетание ионообменной хроматографии с другими методами расширяет область ее применения.
В 1975 г. предложен новый вариант хроматографии, применяемый для определения ионов и названный ионной хроматографией. Для выполнения анализа используют колонки размером 25 Х 0,4 см. Разработана двухколоночная и одноколоночная ионная хроматография. Первая основана на ионообменном разделении ионов на одной колонке с последующим снижением фонового сигнала элюента на второй колонке и кондуктометрическим детектированием, а вторая (без подавления фонового сигнала элюента) сочетается с фотометрическим, атомно-абсорбционным и другими методами детектирования определяемых ионов.
Несмотря на ограниченное число работ по использованию ионной хроматографии в фармацевтическом анализе, очевидна перспективность этого метода для одновременного определения анионного состава многокомпонентных лекарственных форм и солевых растворов для инъекций (содержащих сульфат-, хлорид-, карбонат-, фосфат-ионы), для количественного определения гетероэлементов в органических лекарственных веществах (содержащих галогены, серу, фосфор, мышьяк), для определения уровня загрязнения воды, используемой в фармацевтической промышленности, различными анионами, для определения некоторых органических ионов в лекарственных формах.
Достоинствами ионной хроматографии являются высокая селективность определения ионов, возможность одновременного определен я органических и неорганических ионов, низкий предел обнаружена (до 10-3 и даже 10-6 мкг/мл), малый объем проб и простота их подготовки, быстрота выполнения анализа (за 20 мин возможно разделение до 10 ионов), простота аппаратурного обеспечения, возможность сочетания с другими аналитическими методами и расширение области применения хроматографии в отношении объектов, сходных по химической структуре и трудно разделяемых методами ТСХ, ГЖХ, ЖХВД.
Наиболее широко в фармацевтическом анализе используют хроматографию на бумаге и хроматографию в тонком слое сорбента.
В бумажной хроматографии стационарной фазой служит поверхность специальной хроматографической бумаги. Распределение веществ происходит между водой, находящейся на поверхности бумаги, и подвижной фазой. Последняя представляет собой систему, включающую несколько растворителей.
В фармацевтическом анализе при выполнении испытаний методом бумажной хроматографии руководствуются указаниями ГФ XI, вып. 1 (с. 98) и частных фармакопейных статей на соответствующие лекарственные вещества (лекарственные формы). При испытаниях подлинности хроматографируют на одном листе хроматографической бумаги одновременно испытуемое вещество и соответствующий стандартный образец. Если оба вещества идентичны, то соответствующие им пятна имеют на хроматограммах одинаковый вид и равные значения Rf. Если хроматографировать смесь испытуемого вещества и стандартного образца, то при их идентичности на хроматограмме должно появляться только одно пятно. Чтобы исключить влияние условий хроматографирования на получаемые значения Rf, можно пользоваться более объективной величиной RS, которая представляет собой отношение величин Rf испытуемого и стандартного образцов.
При испытании на чистоту о наличии примесей судят по величине и интенсивности окраски пятен на хроматограмме. Примесь и основное вещество должны иметь разные значения Rf Для полуколичественного определения содержания примеси на одном листе бумаги одновременно в одинаковых условиях получают хроматограмму испытуемого вещества, взятого в определенном количестве, и несколько хроматограмм стандартного образца, взятых в точно отмеренных количествах. Затем сравнивают между собой хроматограммы испытуемого и стандартного образцов. Заключение о количестве примеси делают по величине пятен и их интенсивности.
Количественное содержание вещества методом хроматографии на бумаге можно установить непосредственно на хроматограмме, используя, например, планиметрический, денситометрический, люминесцентный или другие методы. Для количественной оценки используют также способы, основанные на элюировании испытуемого вещества из вырезанного и измельченного участка хроматограммы. В элюате или в сухом остатке (после отгонки экстрагента) содержание испытуемого вещества определяют фотометрическим или электрохимическим методом.
Хроматография в тонком слое сорбента (ГФ XI, вып. 1, с. 102) отличается от хроматографии на бумаге тем, что процесс, протекающий при перемещении подвижной фазы, происходит на носителе (сорбенте), нанесенном тонким слоем на инертную поверхность. Твердый сорбент (неподвижная фаза) может быть закрепленным или не закрепленным на стеклянной пластинке. В качестве сорбента используют силикагель или оксид алюминия (квалификации "для хроматографии"). Для закрепления слоя добавляют небольшие количества сульфата кальция или крахмала. Для ТСХ используют также готовые стандартные пластинки с закрепленным слоем, выпускаемые промышленностью, типа "Силуфол УФ-254" размером 15 Х 15, 20 Х 20 см и др.
Преимуществами ТСХ являются простота приемов и оборудования, высокая чувствительность, широкий набор стандартизованных сорбентов, их устойчивость к температурным и химическим воздействиям, большие потенциальные возможности процессов разделения и детектирования, малая стоимость анализов, возможности проведения испытаний с лекарственными веществами, относящимися к любым классам соединений.
Метод ТСХ широко используется в теоретической и практической фармации для идентификации, обнаружения примесей, количественного определения не только конечных, но и промежуточных продуктов производства лекарств, а также оценки чистоты стандартных образцов лекарственных веществ.
Одним из вариантов ТСХ является хроматография на полиамидных пленках. Преимущества этого варианта заключаются в высокой чувствительности, быстроте выполнения, универсальности, возможности использования различных систем растворителей в малых объемах. Высокую эффективность хроматографического разделения дают такие отечественные сорбенты, как полиамидная крошка марки Б и полиэтилентерефталатная пленка. Подбор системы растворителей осуществляется экспериментальным путем так же, как в ТСХ.
Разработаны различные варианты, позволяющие совершенствовать методы хроматографирования на бумаге и в тонком слое сорбента. В ГФ XI описаны такие специальные приемы, как повторное и двумерное хроматографирование. Они позволяют достигнуть лучшего разделения анализируемых смесей веществ. Суть повторного хроматографирования заключается в том, что полученную хроматограмму высушивают и повторно пропускают подвижную фазу в том же направлении. Двумерное хроматографирование отличается тем, что повторное пропускание той же подвижной фазы осуществляется в перпендикулярном по отношению к первоначальному направлении.
В последние годы разработаны линейная, циркулярная и антициркулярная высокоэффективная тонкослойная хроматографии (ВЭТСХ). Создание последней вызвано получением сорбентов с более узким распределением частиц и пор, а также пленок, почти идеально однородных по толщине. Другие параметры, как, например, среднее значение размеров частиц и ширина их распределения, позволяют сократить время анализа, так как возрастает скорость протекания растворителя между частицами и внутри пор. В качестве сорбентов используют силикагель "Кизельгель 60" с величиной пор 6 нм, целлюлозу и др.
Предложен метод ультрамикрохроматографии. Процесс протекает в микротонком слое специально приготовленного сорбента, что резко повышает скорость и чувствительность анализа.
Для идентификации ряда лекарственных веществ сочетают ТСХ с ИК-спектроскопией, УФ-спектрофотометрией, интерферометрией. Известны два варианта сочетания ТСХ с другими методами для количественного анализа: определение вещества на самой хроматограмме и в элюате (после снятия с хроматограммы). Оба эти варианта очень часто применяют для анализа лекарственных форм. Важные преимущества по сравнению с другими комбинациями физико-химических методов анализа имеет совместное применение ТСХ с денситометрическим определением. Для качественного и количественного анализа фитохимических препаратов и лекарственного сырья используют такие комбинированные методы, как хроматофотометрия, хроматофлуориметрия, хромат о-ИК-спектроскопия, хроматополярография, хроматопланиметрические методы, а также денситофлуориметрия и денситоспектрофотометрия.
Электрофорез на бумаге и в тонких слоях сорбента по технике выполнения и аналитическим возможностям сходен с ТСХ.
В ГФ XI включен электрофорез — метод анализа, основанный на способности заряженных частиц к перемещению в электрическом поле. Подобно ТСХ, электрофорез позволяет разделять и идентифицировать компоненты различных смесей. Скорость перемещения ионов при электрофорезе зависит от напряженности электрического поля, величины заряда, размера частицы, вязкости, рН среды, температуры и других факторов.
Фронтальный электрофорез проводят в свободной незакрепленной фазе в кювете, представляющей собой разборный U-образный канал. В кювете исследуемую смесь и буферный раствор располагают так, чтобы между ними была четкая граница, которая затем в процессе электрофореза расходится на ряд границ, соответствующих числу компонентов.
Зональный электрофорез проводят в закрепленной среде, которая выполняет роль стабилизатора электрофоретических зон. Зональный электрофорез имеет много вариантов: электрофорез в свободной жидкости, на крупнопористых носителях (на бумаге, проточных установках, колонках, в блоке), на мелкопористых носителях (в тонком слое, крахмальном геле, в полиакриламидном геле, диск-электрофорез, изотахофорез).
Известны также комбинированные методы зонального электрофореза — иммуноэлектрофорез и метод пептидных карт (сочетание бумажной и тонкослойной хроматографии с высоковольтным электрофорезом).
Приборы для всех видов электрофореза имеют единую схему. Они содержат камеру для электрофореза, источник тока, электроды, соединяющие камеру с источником тока, и устройства для сбора и идентификации разделяемых веществ. Работа на этих приборах включает такие операции, как подготовка среды, нанесение смеси веществ, проведение электрофореза, обнаружение и количественное определение разделенных веществ.
Оценка полученных результатов электрофореза осуществляется различными способами: зарисовкой или фотографированием, определением величины абсолютной или относительной электрофоретической подвижности, определением физических, химических или биологических показателей каждой фракции, денситометрическим определением. Для окраски электрофореграмм используют красители различного состава или их смеси. Использование в качестве носителя в тонкослойном электрофорезе силигателя марки КСК позволило разработать методики анализа различных лекарственных веществ в таблетках, мазях, эмульсиях, ампулированных растворах, суппозиториях.
Газожидкостная (газовая) хроматография (ГЖХ) основана на распределении вещества между газовой и жидкой или твердой фазами.
Преимущество ГЖХ перед другими хроматографическими методами заключается в универсальности (можно разделять смеси газов, жидкостей и твердых веществ); способности разделять сложные смеси, содержащие до нескольких десятков компонентов; короткой продолжительности анализа (5—20 мин); достаточно высокой чувствительности (до 10-13 г); малой величине анализируемой пробы (до 10-4 г); сравнительно небольшой относительной ошибке (1—1,5%), которая может быть уменьшена (до 0,01—0,02%) при использовании интеграторов; простоте конструкции и надежности эксплуатации газовых хроматографов, Эти достоинства способствовали активному внедрению ГЖХ в практику анализа лекарственных веществ с различными физическими свойствами.
Прибор для ГЖХ — газовый хроматограф — включает систему измерения и регулирования скорости потока газа-носителя, систему ввода пробы испытуемого образца, газохроматографическую колонку, систему термостатирования и контроля температуры в различных узлах прибора и систему детектирования, регистрации и обработки полученной на приборе информации.
Газ-носитель поступает в хроматограф из баллона через редуктор. Система ввода анализируемой пробы включает испаритель и мембрану из термостойкой резины, расположенной на вводе. Объем пробы жидкости составляет 0,1—1 мкл, а газа — от 0,5 до 5 мл. Колонка для ГЖХ представляет собой трубку (прямую или спиральную), изготовленную из нержавеющей стали или стекла, с внутренним диаметром 0,6—5 мм и длиной от 1 до 3 м. Твердый носитель служит для удерживания тонкой пленки неподвижной жидкой фазы. Готовят твердые Носителе из материалов на основе кремнезема — диатомита или ки- зельгура, фтор углеродных полимеров, полистирола и др. В качестве неподвижной жидкой фазы используют высококипящую жидкость — это углеводороды или их смеси, простые и сложные эфиры, полифенолы, амины, жирные кислоты и др. Испытуемое вещество (смесь) вводят в поток газа-носителя, где оно испаряется, и в виде пара проходит через колонку с сорбентом, распределяясь между газовой и жидкой или газовой и твердой фазами. Разделенные вещества элюируются из колонки газом-носителем, регистрируются детектором и фиксируются на хроматограмме в виде пиков. Наиболее часто используют детектор по теплопроводности и пламенно-ионизационный, а также термоионный и электронозахватный. Действие детектора основано на измерении теплопроводности газа-носителя в присутствии других веществ, а пламенно-ионизационного — на измерении тока насыщения ионизированной газовой смеси в зависимости от ее состава. Термоионный и электронозахватный детекторы более селективны и чувствительны.
Метод ГЖХ может быть использован для испытаний подлинности лекарственных веществ. С этой целью применяют либо способ, основанный на использовании веществ-свидетелей, либо метод относительных удерживаний. В первом случае после анализа исследуемого образца в идентичных условиях хроматографируют вещества-свидетели, которые могут содержаться в испытуемом объекте. Если времена удерживания свидетеля и какого-либо компонента в испытуемом образце совпадают, то это может служить доказательством идентичности данных веществ. При испытании подлинности методом относительных удерживаний к пробе прибавляют определенное количество вещества-свидетеля. Затем анализируют по рекомендуемой методике и рассчитывают по формуле величину относительного удерживания, которая является постоянной для вещества в конкретных условиях выполнения анализа.
Количественный ГЖХ анализ лекарственных веществ выполняют в тех же условиях, что и качественный. Для расчетов количественного содержания используют такие параметры, как площадь или высота пиков веществ. Площадь пика на хроматограмме можно установить с помощью планиметра, интегратора или умножением высоты пика на его полуширину (ширину, измеренную на половине высоты).
Для количественного ГЖХ анализа используют такие методы, как метод абсолютной градуировки, метод внутренней нормализации и метод внутреннего стандарта. Сущность метода абсолютной градуировки заключается в установлении зависимости между количеством введенного в хроматограф вещества и высотой (или площадью) пика. При использовании метода внутренней нормализации сумму площадей пиков приводят к 100%, а затем вычисляют содержание каждого из них. Применение метода внутреннего стандарта основано на сравнении высоты или площади пика анализируемого и стандартного вещества, введенного в пробу в определенном количестве.
Метод ГЖХ все шире используется для качественного анализа лекарственных средств. На различных сорбентах величины относительного удерживания установлены для лекарственных веществ из различных химических групп, в том числе альдегидов, кетонов, фенолов, терпеноидов, амидов и сложных эфиров карбоновых кислот, амино- производных, производных пиразола, имидазола, барбитуровой кислоты, некоторых алкалоидов, а также для ряда сильнодействующих лекарственных веществ из числа производных фенилалкиламинов, бензодиазепина, фенотиазина (Н.Н.Дементьева).
Газовую хроматографию сочетают с другими методами. Для анализа настоек, представляющих собой тройные системы, применяют рефрактохроматографический метод. Сущность его заключается в том, что соотношение концентрации спирта и воды устанавливают методом ГЖХ, а экстрактивные вещества — по показателю преломления. Эффективным оказалось сочетание ГЖХ и масс-спектрометрии. Хромато-масс-спектрометрические характеристики позволили осуществить качественный и количественный анализ ряда препаратов. Широкое распространение приобретает один из вариантов ГЖХ — капиллярная хроматография, основанная на использовании колонок диаметром 0,1—1,0 мм и длиной 50—100 м.
Жидкостная хроматография (ЖХ) отличается от газовой хроматографии тем, что подвижной фазой служит не газ, а жидкость. В зависимости от характера неподвижной фазы различают твердожидкостную и жидко-жидкостную хроматографию. Вариантом колоночной ЖХ является высокоэффективная жидкостная хроматография (ВЭЖХ), которую называют также жидкостной хроматографией высокого давления. Характерной особенностью ВЭЖХ является то, что подвижная фаза проходит через колонку, наполненную сорбентом с большой скоростью за счет значительного давления. Метод ВЭЖХ позволяет разделять на индивидуальные вещества многокомпонентные смеси нелетучих органических соединений сложной химической структуры с различной молекулярной массой. Метод очень широко используется для идентификации и количественного определения в аналитической химии и в фармацевтическом анализе. Чувствительность ВЭЖХ достигает 10-6 г. На разделение смеси из 10—15 компонентов затрачивается 20—30 мин, причем выделяются вещества высокой степени чистоты.
Жидкостный хроматограф включает такие узлы, как дозатор, насос высокого давления, высокоэффективная колонка, детектор с регистрирующим устройством. Современные приборы оснащены устройствами, позволяющими автоматически вводить пробу, с помощью микропроцессора выполнять заданную программу хроматографического процесса, автоматически оптимизировать условия разделения и выдавать результаты, позволяющие осуществить качественную и количественную оценку анализируемой смеси веществ.
Подача элюента в колонку с заданной скоростью осуществляется с помощью насоса высокого давления до 20—50 МПа (200—500 атм) или низкого давления до 1—2 МПа (10—20 атм). Колонки для хроматографирования изготавливают из нержавеющей стали. Они имеют длину 10—25 см, внутренний диаметр 0,3—0,8 см и заполняются адсорбентом с диаметром частиц 5—10 мкм (сферической или неправильной формы). Адсорбент плотно упаковывается, что позволяет достигнуть высокоэффективного разделения смеси. Разделение производят в интервале температур 20—50° С, поддерживая ее с точностью ±0,1° С.
В приборах для ВЭЖХ используют спектрофотометрический, рефрактометрический, флуориметрический, пламенно-ионизационный, масс-спектрометрический, электрохимический и другие детекторы. Чаще всего детектором является спектрофотометр с переменной (190—900 нм) длиной волны.
Адсорбентом обычно служит силикагель либо с гидроксилированной поверхностью, либо с привитыми к поверхности различными функциональными группами. В качестве элюента используют различные углеводороды, добавляя к ним небольшие количества этанола или других растворителей. В обращенно-фазной ВЭЖХ колонки заполняют силикагелем с привитыми гидрофобными группами, а в качестве элюента берут водные растворы низших спиртов или ацетонитрил. В тех же условиях применяют ионпарную ВЭЖХ для разделения органических кислот, оснований и их солей, но в этом случае к элюенту добавляют ионные соединения, анион или катион которых содержит гидрофобную группу. Ионообменную ВЭЖХ применяют для разделения органических катионов и анионов, используя в качестве адсорбентов соединения, содержащие сульфо-группы, карбоксильные или аминогруппы разной основности. Элюентами служат водные буферные растворы с различным рН.
Вещества, способные образовывать комплексы с катионами металлов (например, оптические изомеры аминокислот), разделяют с помощью лигандообменной ВЭЖХ. Адсорбентами при этом служат соединения, способные образовывать комплексы с ионами металлов и разделяемым веществом.
Одной из разновидностей метода является микроколоночная жидкостная хроматография, представляющая собой универсальный ультрачувствительный и высокоэкономичный вариант ВЭЖХ. Принципиальное его отличие заключается в использовании микроколонок объемом около 200 мкл и спектрофотометра с объемом проточной кюветы 1 мкл в качестве детектора. В Российской Федерации выпускается микроколоночный жидкостный хроматограф "Милихром".
Метод ВЭЖХ успешно применен для качественной и количественной оценки ряда наркотических, ядовитых и сильнодействующих лекарственных веществ. С этой целью перспективно использование таких параметров, как время удерживания, коэффициент емкости, интегральные спектральные отношения (А.Х.Лайпанов). В нашей стране метод ВЭЖХ рекомендован во всех ФС, разработанных на стандартные образцы антибиотиков. В Фармакопее США (XII издание) метод ВЭЖХ применен для анализа большинства антибиотиков противоопухолевого действия и цефалоспоринового ряда.
Ряд преимуществ по сравнению с ГЖХ и ВЭЖХ имеет занимающая промежуточное положение между этими методами сверхкритическая флюидная хроматография. Преимущества метода заключаются в возможности разделения неустойчивых и нелетучих веществ, универсальном детектировании и высокой экспрессности. Так, например, сочетание сверхкритической флюидной хроматографии с УФ-детектором позволяет детектировать 40 алкалоидов в одном растении.
Эксклюзионная хроматография (гель-фильтрация, молекулярно-ситовая фильтрация, гельпроникающая хроматография) — вид жидкостной распределительной хроматографии, в процессе которой разделение происходит по размерам молекул. В гель-фильтрации элюентом служит вода, а в гельпроникающей хроматографии — органический растворитель. Неподвижной фазой являются пористые материалы с определенным узким распределением пор по диаметру. Исследуемое вещество, диаметр которого превышает диаметр пор, не может диффундировать внутрь таких сорбентов. Поэтому оно проходит через колонку быстрее, чем молекулы с меньшим размером, которые проникают в поры. Таким образом обеспечивается разделение молекул в зависимости от их размера. Поры внутри геля заполнены жидкостью, которая занимает большую часть его объема. Применяют гели на основе декстрана, агарозы, полиакриламида. Метод может быть использован для определения молекулярной массы, а также для исследования, очистки и разделения веществ с молекулярной массой 102—108. Осуществляют эксклюзионную хроматографию в жидкостных хроматографах.
Колонки заполняют различными сорбентами: мягкими (сефадексы), полужесткими (полиакриламидные гели), жесткими (пористые стекла). Детектором служит проточный рефрактометр или спектрофотометр. В отличие от других видов хроматографии на выполнение испытания требуются небольшие затраты времени.
Количественное содержание каждого компонента смеси можно установить используя метод внутреннего стандарта или абсолютной калибровки, т.е. так же, как в ГЖХ. Время выхода каждого компонента из колонки в идентичных условиях разделения является постоянной величиной и может служить для идентификации вещества. Площадь пика пропорциональна количеству компонента и поэтому используется для определения его содержания в смеси.
Полибуферное распределение в фармацевтическом анализе применяют для разделения смесей веществ. Оно может быть использовано в анализе лекарственных форм, представляющих смеси оснований или кислот, для разделения смесей аминов, алкалоидов, органических кислот, фенолов, антибиотиков, а также для отделения веществ, диссоциирующих на ионы, от не диссоциирующих.
Экстракцию как метод разделения применяют в фармацевтическом анализе, особенно для разделения компонентов, входящих в состав лекарственных форм. В зависимости от исходной фазы различают экстракцию из твердого вещества и экстракцию из раствора (жидкостную), а по количеству операций — однократную и многократную экстракции. Основное условие разделения — выбор экстрагента, не смешивающегося с исходной фазой и легко отделяющегося от нее и от экстрагируемого вещества. Экстракцию как метод разделения сочетают с фотометрией.
Экстракционно - фотометрический метод основан на образовании цветных продуктов, способных экстрагироваться каким-либо органическим растворителем. Этот метод используют для анализа многих препаратов и лекарственных форм. Метод включен в ГФ XI, зарубежные фармакопеи.
Для экстракционно-фотометрического определения алифатических, ароматических, гетероциклических азотсодержащих лекарственных веществ используют различные группы кислотных красителей: азокрасители (метиловый оранжевый, магнезон ИРЕА, кислотный хром темно-синий, тропеолин 00), сульфофталеиновые красители (бромфеноловый синий, бромтимоловый синий, бромкрезоловый зеленый, бромкрезоловый пурпуровый, тимоловый синий, пирокатехиновый фиолетовый, ксиленоловый оранжевый), оксиксантеновые красители (эозин, эритрозин, флоксин, бенгальский розовый А и Б). Указанные красители образуют с азотсодержащими соединениями и их солями окрашенные комплексы или ионные ассоциаты, растворы которых отличаются высокими значениями молярных коэффициентов поглощения, что позволяет определять малые количества веществ. Окрашенные вещества экстрагируют в органическую фазу (обычно в хлороформ) и измеряют оптическую плотность с помощью спектрофотометра или фотоколориметра.
Чаще всего измеряют оптическую плотность раствора ионного ассоциата в органическом растворителе. Но в ряде случаев полученный ассоциат разрушают введением кислоты в органическую фазу или путем реэкстракции красителя водными растворами кислот или оснований, а затем измеряют абсорбцию свободного красителя. Анализ выполняют при оптимальном значении рН водной среды, которую устанавливают экспериментально для каждого испытуемого препарата. Наряду со стехиометрическим вариантом экстракционно-фотометрического метода применяют также субстехиометрический, сущность которого состоит в однократном экстрагировании ионного ассоциата, что в значительной степени упрощает методику выполнения анализа и сокращает время его выполнения.
Аминопроизводные соединения алифатического, ароматического и гетероциклического ряда ввиду наличия неподеленной пары электронов атома азота имеют высокую реакционную способность. Они, в частности, вступают в реакции с комплексными металлокислотами.
В качестве реактива, образующего тройные комплексы: органическое основание — таллий (III) — галогенид с препаратами, содержащими в молекуле третичный атом азота, и четвертичными аммониевыми основаниями, — был использован тетрабромид таллия (III) — бромталлиевая кислота (Г.И.Олешко). Разработаны способы экстракционно-фотометрического определения производных арилалифатических соединений (димедрол, спазмолитин), гетероциклических (акрихин, тропацин, гидрохлориды кокаина и папаверина), четвертичные аммониевые соединения (котарнина хлорид).
Тройные комплексы, образованные тиоцианатом железа (III) и молибдена (V) с алкалоидами и другими органическими основаниями, экстрагируются хлороформом в среде 2—3 М соляной кислоты, а тройные комплексы с пирокатехинатом молибдена (VI) — при рН 2. Это послужило основой для разработки способов экстракционно-фотометрического определения в лекарственных формах димедрола, дибазола, антипирина с тиоцианатом молибдена (V), папаверина гидрохлорида с тиоцианатом железа (III), промедола и кокаина гидрохлорида с пирокатехинатом молибдена (VI) (С.Г.Дуксина).
Разрабатываются или получают дальнейшее развитие другие методы разделения, например эксорбция, представляющая собой сочетание экстракции и сорбции. Метод более эффективно позволяет извлекать растворенное вещество по сравнение с экстракционным или адсорбционным процессом. Большие возможности открывает использование капиллярного изотахофореза с УФ-детекторами в анализе различных природных биологически активных веществ. Метод дает возможность идентифицировать, устанавливать степень чистоты, определять количественно и испытывать стабильность лекарственных веществ, а также анализировать двух- и трехкомпонентные лекарственные формы.
Проточно - инжекционный анализ отличается простотой и доступностью аппаратуры, высокой производительностью (100—200 анализов/ч), возможностью широкого варьирования анализируемых концентраций. Особенно перспективен этот метод в серийном анализе и в разных точках технологических линий. Для детектирования используют фотометрические, амперометрические, флуориметрические методы.
4.8 Термические методы анализа
Нагревание лекарственных веществ до температуры, не вызывающей термического разложения, приводит к ряду изменений в их физических свойствах. Происходят полиморфные превращения, растворение в кристаллизационной воде, удаление сорбционной и кристаллизационной воды, сублимация, плавление, кипение. В зависимости от природы вещества, температуры и условий нагревания могут происходить химические превращения: структурирование, термическая, окислительная или гидролитическая деструкция. Термическая деструкция веществ сопровождается поглощением или выделением теплоты, а также образованием газообразных продуктов. Поэтому наиболее информативными и экспрессными методами оценки термической стабильности являются термография и термогравиметрия.
Термография позволяет оценить термическую стабильность по температурам термоэффекта, связанного с деструкцией исследуемого вещества.
Термогравиметрия дает возможность определить термическую стабильность по температуре, при которой наблюдается уменьшение массы вещества.
Термографический анализ лекарств весьма перспективен. Специфичность термограмм позволила использовать метод для идентификации целого ряда лекарственных веществ.
Характерная особенностьтермометрического титрования — его универсальность. Один и тот же прибор можно использовать для различных видов определений, для анализа не требуются специфические индикаторы, селективные электроды. Область применения метода — реакции нейтрализации, окисления — восстановления и др.
Термический анализ основан на точной (до 0,1°С) регистрации равновесного состояния между кристаллической и жидкой фазами анализируемого вещества. Медленно нагревая или охлаждая сплав, устанавливают температуру, при которой появляются и исчезают кристаллы. Модификацией термического анализа является термомикроскопический метод, в котором используется поляризационная микроскопия для установления фазовых равновесий. Основные недостатки этих методов: невозможность использования для исследования термолабильных веществ, значительные затраты времени на выполнение анализа, отсутствие должной воспроизводимости. Значительно лучшей воспроизводимостью отличается дифференциальный термический анализ, основанный на регистрации изменения энергии в зависимости от температуры. Дифференциальный термический анализ позволяет определять наличие примесей в лекарственных веществах, прогнозировать сроки их годности, устанавливать однородность каждой партии и т.д.
Одной из модификаций дифференциального термического анализа, используемых для получения термических характеристик веществ, является дериватография. Сущность дериватографии заключается в регистрации изменений температуры образца, вызванных дегидратацией, плавлением, термической деструкцией и другими процессами, происходящими при его непрерывном нагревании. Метод сочетает экспрессность и информативность. С помощью дериватографов можно одновременно автоматически регистрировать простую и дифференциальную кривые нагревания, кривые и скорости изменения массы как функции времени. Используя указанные характеристики, можно исследовать полиморфные структуры, идентифицировать лекарственные вещества, давать качественную оценку стандартным образцам, изучать кинетику сушки и определять содержание влаги в лекарственных веществах или промежуточных продуктах их получения.
Дериватография оказалась эффективным методом для определения содержания влаги и общей золы в лекарственном растительном сырье, а также определения сухого остатка в жидких лекарственных формах, получаемых из этого сырья (настойки и экстракты).
Один из вариантов дифференциального термического анализа — дифференциальная сканирующая калориметрия, применение которой в комплексе с другими физико-химическими методами оказалось эффективным для оценки качества стандартных образцов, отличающихся высокой степенью чистоты.
Метод дифференциальной микрокалориметрии, основанный на определении энтальпии плавления, рекомендован для количественного определения термически неустойчивых веществ, установления степени чистоты, стабильности, наличия полиморфных форм.
Термофрактография — метод, основанный на нагревании сырья в определенном температурном интервале и улавливании образующихся продуктов по фракциям.
В последние годы проводятся исследования по комплексному применению физических и физико-химических методов. Это обеспечивает возможность получения новых характеристик и констант, позволяющих дать всестороннюю оценку лекарственного вещества или группы препаратов сходной химической структуры.
Глава 5. Биологические методы анализа 5.1 Биологический контроль качества лекарственных средств
Биологическую оценку качества лекарственных препаратов обычно проводят по силе фармакологического эффекта или по токсичности. Применяют биологические методы, когда с помощью физических, химических или физико-химических методов не удается сделать заключение о чистоте или токсичности лекарственного препарата или когда способ получения препарата не гарантирует постоянства активности (например, антибиотики).
Проводят биологические испытания на животных (кошки, собаки, кролики, лягушки и др.), отдельных изолированных органах (рог матки, часть кожи), отдельных группах клеток (форменные элементы крови), а также на определенных штаммах микроорганизмов. Активность препаратов выражают в единицах действия (ЕД).
Биологический контроль лекарств, содержащих сердечные гликози- ды. По ГФ XI проводят биологическую оценку активности лекарственного растительного сырья и полученных из него препаратов, содержащих сердечные гликозиды, в частности наперстянки (пурпурной, крупноцветковой и шерстистой), горицвета, ландыша, строфанта, желтушника серого. Испытания проводят на лягушках, кошках и голубях, устанавливая соответственно лягушачьи (ЛЕД), кошачьи (КЕД) и голубиные (ГЕД) единицы действия. Одна ЛЕД соответствует дозе стандартного образца, вызывающего в условиях опыта систолическую остановку сердца у большинства подопытных стандартных лягушек (самцы массой 28—33 г). Одна КЕД или ГЕД соответствует дозе стандартного образца или испытуемого препарата из расчета на 1 кг массы животного или птицы, вызывающего систолическую остановку сердца кошки или голубя. Содержание ЕД рассчитывают в 1,0 г исследуемого лекарственного средства, если испытывают растительное сырье или сухие концентраты; в одной таблетке или в 1 мл, если испытывают жидкие лекарственные формы.
Испытание на токсичность. В этот раздел ГФ XI, вып. 2 (с. 182) по сравнению с ГФ X внесен ряд дополнений и изменений, отражающих возрастающие требования к качеству лекарственных средств и необходимость унификации условий их испытаний. В статью введен раздел, в котором описан порядок отбора проб. Увеличена масса животных, на которых проводят испытание, указаны условия их содержания и срок наблюдения за ними. Для выполнения испытания отбирают по два флакона или ампулы от каждой серии, содержащей не более 10000 флаконов или ампул. Из партий с большим количеством отбирают по три ампулы (флакона) от каждой серии. Содержимое проб одной серии смешивают и испытывают на здоровых белых мышах обоего пола массой 19—21 г. Испытуемый раствор вводят в хвостовую вену пяти мышей и ведут наблюдение за животными 48 ч. Препарат считается выдержавшим испытание, если ни одна из подопытных мышей не погибнет в течение указанного срока. В случае гибели даже одной мыши испытание повторяют по определенной схеме. В частных статьях может быть указан и другой порядок проведения испытания на токсичность.
Испытания на пирогенность. Бактериальные пирогены представляют собой вещества микробного происхождения, способные вызвать у человека и теплокровных животных при попадании в кровяное русло повышение температуры тела, лейкопению, падение кровяного давления и другие изменения в различных органах и системах организма. Пирогенную реакцию вызывают грамотрицательные живые и мертвые микроорганизмы, а также продукты их распада. Допустимо содержание, например, в изотоническом растворе натрия хлорида 10 микроорганизмов в 1 мл, а при введении не более 100 мл допускается 100 на 1 мл. Испытанию на пирогенность подвергают воду для инъекций, инъекционные растворы, иммунобиологические лекарственные средства, растворители, используемые для приготовления инъекционных растворов, а также лекарственные формы, вызывающие, по сведениям клиник, пирогенную реакцию.
В ГФ XI, как и в фармакопеи других стран мира, включен биологический метод испытания пирогенности, основанный на измерении температуры тела кроликов после введения в ушную вену испытуемых стерильных жидкостей. Отбор проб проводится так же, как при испытании на токсичность. В общей статье (ГФ XI, вып. 2, с. 183—185) указаны требования к подопытным животным и порядок их подготовки к проведению испытаний. Испытуемый раствор проверяют на трех кроликах (не альбиносах), масса тела которых отличается не более чем на 0,5 кг. Температуру тела измеряют, вводя термометр в прямую кишку на глубину 5—7 см. Испытуемые жидкости считают непирогенными, если сумма повышенной температуры у трех кроликов равна или меньше 1,4°С. Если эта сумма превышает 2,2°С, то воду для инъекций или инъекционный раствор считают пирогенными. Если сумма повышения температуры у трех кроликов находится в пределах от 1,5 до 2,2° С, испытание повторяют дополнительно на пяти кроликах. Испытуемые жидкости считают непирогенными, если сумма повышений температуры у всех восьми кроликов не превышает 3,7°С. В частных ФС могут быть указаны другие пределы отклонений температуры. Кроликов, бывших в опыте, можно использовать для этой цели повторно не ранее чем через 3 сут., если введенный им раствор был непирогенным. Если же введенный раствор оказался пирогенным, то кроликов повторно можно использовать только через 2—3 недели. В ГФ XI по сравнению с ГФ X введена проверка на реактивность кроликов, впервые используемых для испытаний, и уточнен раздел о возможности их использования для повторных испытаний.
Рекомендуемый ГФ XI биологический метод отличается специфичностью, но не дает количественной оценки содержания пирогенных веществ. К существенным его недостаткам следует отнести трудоемкость и продолжительность испытаний, необходимость содержания животных, ухода за ними, сложность подготовки к проведению испытаний, зависимость результатов от индивидуальных особенностей каждого животного и т.д. Поэтому предпринимались попытки разработки других методов определения пирогенности.
Наряду с определением пирогенности на кроликах за рубежом используют микробиологический метод, основанный на подсчете общего числа микроорганизмов в исследуемой лекарственной форме до ее стерилизации. В нашей стране предложена простая и доступная методика обнаружения пирогенов, основанная На избирательной идентификации грамотрицательных микроорганизмов по реакции образования геля с применением 3%-ного раствора гидроксида калия. Методика может быть использована на химико-фармацевтических предприятиях.
Предпринята попытка заменить биологический метод определения пирогенности химическим. Растворы, содержащие пирогены, после обработки хиноном показывали отрицательную реакцию с тетрабромфенолфталеином. Пирогенал с триптофаном в присутствии серной кислоты образует буро-малиновое окрашивание при содержании пирогенала 1 мкг и более.
Исследовалась возможность спектрофотометрического определения пирогенных веществ в УФ-области спектра. Растворы фильтрата пирогенсодержащих культур микроорганизмов обнаруживают слабовыраженный максимум поглощения при 260 нм. По чувствительности спектрофотометрический метод определения пирогенов в 7-8 раз уступает биологическому испытанию на кроликах. Однако если перед спектрофотометрированием провести ультрафильтрование, то вследствие концентрирования пирогенов можно достигнуть сопоставимых результатов определения биологическим и спектрофотометрическим методами.
После обработки хиноном растворы пирогенов приобретают красную окраску и появляется максимум светопоглощения при 390 нм. Это позволило разработать фотоколориметрический способ определения пирогенов.
Высокая чувствительность люминесцентного метода создала предпосылки использования его для определения пирогенных веществ в концентрации до 1*10-11 г/мл. Разработаны методики люминесцентного обнаружения пирогенов в воде для инъекций и в некоторых инъекционных растворах с применением красителей родамина 6Ж и 1-анилино-нафталин-8-сульфоната. Методики основаны на способности пирогенов увеличивать интенсивность люминесценции указанных красителей. Они позволяют получать результаты, сопоставимые с биологическим методом.
Относительная ошибка спектрофотометрического и люминесцентного определения не превышает ±3%. Для определения пирогенности воды для инъекций используют также хемилюминесцентный метод.
Перспективным методом является полярография. Установлено, что фильтраты пирогенных культур даже в очень разбавленном состоянии оказывают сильное подавляющее действие на полярографический максимум кислорода. На этой основе разработан способ полярографической оценки качества воды для инъекций и некоторых инъекционных растворов.
Испытание на содержание веществ гистаминоподобного действия.
Данному испытанию подвергают парентеральные лекарственные средства. Выполняют его на кошках обоего пола массой не менее 2 кг под уретановым наркозом. Вначале животному, находящемуся под наркозом, вводят гистамин, проверяя его чувствительность к этому веществу. Затем с интервалом 5 мин продолжают повторные введения (0,1 мкг/кг) стандартного раствора гистамина до тех пор, пока при двух последовательных введениях не будет получено одинаковое снижение артериального давления, которое принимается за стандартное. После этого с интервалом 5 мин животному вводят испытуемый раствор с той же скоростью, с которой вводили гистамин. Препарат считают выдержавшим испытание, если снижение артериального давления после введения тест-дозы не превышает реакции на введение 0,1 мкг/кг в стандартном растворе.
5.2 Микробиологический контроль лекарственных средств
Впервые в ГФ XI включен новый вид биологического контроля — определение микробиологической чистоты нестерильных лекарственных средств, т.е. установление состава и количества имеющейся в препарате микрофлоры и ее соответствие нормам, ограничивающим микробную обсемененность (контаминацию). Патогенные микроорганизмы (синегнойная палочка, кишечные бактерии) способны находиться в таблетках и гранулах от 6 до 18 мес., сохраняя морфологические и биохимические свойства. Микробиологическая чистота нестерильных лекарственных средств находится в зависимости от санитарно-гигиенических условий производства, дополнительной обработки сырья с целью его деконтаминации и состояния микробиологического контроля ОТК на всех этапах производства.
Испытание на микробиологическую чистоту. Необходимость проведения этого испытания вызвана тем, что лекарственные средства, в том числе выпускаемые в виде таких лекарственных форм, как таблетки, капсулы, гранулы, растворы, сиропы, мази, не стерилизуются в процессе производства. Поэтому они могут быть загрязнены микроорганизмами. Испытание включает количественное определение жизнеспособных бактерий и грибов и выявление некоторых видов микроорганизмов, представителей кишечной флоры и стафилококка, содержание которых недопустимо в нестерильных лекарственных средствах.
Выполняют испытание на микробиологическую чистоту в асептических условиях. В общей статье подробно описаны методы и питательные среды для контроля всех видов нестерильных лекарственных средств (ГФ XI, вып. 2, с. 193). Количественное определение микроорганизмов выполняют двухслойным агаровым методом в чашках Петри. Образец лекарственного средства в количестве 10 г (мл) растворяют, суспендируют или эмульгируют в фосфатном буферном растворе (рН 7,0) с таким расчетом, чтобы конечный объем раствора был 100 мл. Затем по 1 мл образца смешивают с питательной средой (4 мл).
Через 5 сут инкубирования при 30—35° С подсчитывают число бактериальных колоний на двух чашках и вычисляют число бактерий в 1 г (мл) образца.
Испытания на стерильность. Целью этого испытания является доказательство отсутствия в лекарственном средстве жизнеспособных микроорганизмов любого вида с максимально возможной достоверностью. Результаты испытания на стерильность — один из важнейших показателей безопасности лекарственных средств.
Этому испытанию подвергаются все лекарства для парентерального введения, глазные капли и мази, лекарства, наносимые на открытую рану, и др. Эффективность данного вида контроля определяют такие факторы, как отбор образца для анализа, техника посева лекарственного средства, состав питательных сред, время инкубации йосевов, интерпретация и учет результатов испытания.
Приведенные в общей статье на стерильность (ГФ XI, вып.2, с. 187) методы контроля применяют для испытаний всех лекарственных средств независимо от их химической природы и лекарственной формы, если нет других указаний в частных статьях.
Для установления стерильности лекарства вначале определяют его антимикробное действие. Для контроля стерильности применяют биогликолевую среду и жидкую среду Сабуро, используя при этом метод прямого посева на питательные среды. Если лекарственное средство обладает выраженным антимикробным действием или разлито в емкости более 100 мл, то для контроля его стерильности используют метод мембранной фильтрации. Испытание выполняют на фильтрационной установке, включающей фильтродержатель с мембранным фильтром и колбу-приемник.
Метод мембранной фильтрации включен во многие зарубежные фармакопеи, как основной для проведения стерилизации. Несмотря на отдельные недостатки, этот метод имеет целый ряд преимуществ в отличие от прямого посева. Он устраняет антимикробное действие препаратов, которое при прямом посеве может исказить результаты определения, дает возможность исследовать большие объемы лекарственного средства, сократить время инкубации посевов, экономить питательные среды и т.д.
Выводы
Одна из наиболее важных задач фармацевтической химии — это разработка и совершенствование методов оценки качества лекарственных средств.
Для установления чистоты лекарственных веществ используют различные физические, физико-химические, химические методы анализа или их сочетание. ГФ предлагает следующие методы контроля качества ЛC.
Физические и физико-химические методы. К ним относятся:
· определение температур плавления и затвердевания, а также температурных пределов перегонки;
· определение плотности,
· определение показателей преломления (рефрактометрия),
· определение оптического вращения (поляриметрия);
· спектрофотометрия (ультрафиолетовая, инфракрасная);
· фотоколориметрия,
· эмиссионная и атомно-абсорбционная спектрометрия,
· флуориметрия,
· спектроскопия ядерного магнитного резонанса,
· масс-спектрометрия;
· хроматография (адсорбционная, распределительная, ионообменная, газовая, высокоэффективная жидкостная);
· электрофорез (фронтальный, зональный, капиллярный);
· электрометрические методы (потенциометрическое определение рН, потенциометрическое титрование, амперометрическое титрование, вольтамперометрия).
Кроме того, возможно применение методов, альтернативных фармакопейным, которые иногда имеют более совершенные аналитические характеристики (скорость, точность анализа, автоматизация). В некоторых случаях фармацевтическое предприятие приобретает прибор, в основе использования которого лежит метод, еще не включенный в Фармакопею (например, метод рамановской спектроскопии — оптический дихроизм). Иногда целесообразно при определении подлинности или испытании на чистоту заменить хроматографическую методику на спектрофотометрическую. Фармакопейный метод определения примесей тяжелых металлов осаждением их в виде сульфидов или тиоацетамидов обладает рядом недостатков. Для определения примесей тяжелых металлов многие производители внедряют такие физико-химические методы анализа, как атомно-абсорбционная спектрометрия и атомно-эмиссионная спектрометрия с индуктивно связанной плазмой.
Важной физической константой, характеризующей подлинность и степень чистоты ЛC, является температура плавления. Чистое вещество имеет четкую температуру плавления, которая изменяется в присутствии примесей. Для веществ, которые плавятся с разложением, обычно указывается температура, при которой вещество разлагается и происходит резкое изменение его вида.
В некоторых частных статьях ГФ X рекомендуется определять температуру затвердевания или температуру кипения (по ГФ XI — "температурные пределы перегонки") для ряда жидких ЛC. Температура кипения должна укладываться в интервал, приведенный в частной статье. Более широкий интервал свидетельствует о присутствии примесей.
Во многих частных статьях ГФ X приведены допустимые значения плотности, реже вязкости, подтверждающие подлинность и доброкачественность ЛC.
Практически все частные статьи ГФ X нормируют такой показатель качества ЛC, как растворимость в различных растворителях. Присутствие примесей в ЛB может повлиять на его растворимость, снижая или повышая ее в зависимости от природы примеси.
Критериями чистоты являются также цвет ЛB и/или прозрачность жидких лекарственных форм.
Определенным критерием чистоты JIC могут служить такие физические константы, как показатель преломления луча света в растворе испытуемого вещества (рефрактометрия) и удельное вращение, обусловленное способностью ряда веществ или их растворов вращать плоскость поляризации при прохождении через них плоскополяризованного света (поляриметрия). Методы определения этих констант относятся к оптическим методам анализа и применяются также для установления подлинности и количественного анализа ЛС и их лекарственных форм.
Важным критерием доброкачественности целого ряда ЛС является содержание в них воды. Изменение этого показателя (особенно при хранении) может изменить концентрацию действующего вещества, а, следовательно, и фармакологическую активность и сделать ЛС не пригодным к применению.
Химические методы. К ним относятся: качественные реакции на подлинность, растворимость, определение летучих веществ и воды, определение содержания азота в органических соединениях, титриметрические методы (кислотно-основное титрование, титрование в неводных растворителях, комплексонометрия), нитритометрия, кислотное число, число омыления, эфирное число, йодное число и др.
Биологические методы. Биологические методы контроля качества ЛС весьма разнообразны. Среди них испытания на токсичность, стерильность, микробиологическую чистоту.
Для проведения физико-химического анализа полупродуктов, субстанций лекарственных средств и готовых лекарственных форм при проверке их качества на соответствие требованиям ФС контрольно-аналитическая лаборатория должна быть оснащена следующим минимальным набором оборудования и приборов:
· ИК-спектрофотометр (для определения подлинности); спектрофотометр для спектрометрии в видимой и УФ-области (определение подлинности, количественное определение, однородность дозирования, растворимость);
· оборудование для тонкослойной хроматографии (ТСХ) (определение подлинности, родственных примесей);
· хроматограф для высокоэффективной жидкостной хроматографии (ВЭЖХ) (определение подлинности, количественное определение, определение родственных примесей, однородности дозирования, растворимости);
· газожидкостной хроматограф (ГЖХ) (содержание примесей, определение однородности дозирования);
· поляриметр (определение подлинности, количественное определение);
· потенциометр (измерение рН, количественное определение);
· атомно-абсорбционный спектрофотометр (элементный анализ тяжелых металлов и неметаллов);
· титратор К. Фишера (определение содержания воды);
· дериватограф (определение потери массы при высушивании).
Список использованной литературы
1. Арзамасцев А.П. Фармакопейный анализ – М.: Медицина, 1971.
2. Беликов В.Г. Фармацевтическая химия. В 2 частях. Часть 1. Общая фармацевтическая химия: Учеб. для фармац. ин-тов и фак. мед. ин-тов. — М.: Высш. шк., 1993. - 432 с.
3. Глущенко Н. Н. Фармацевтическая химия: Учебник для студ. сред. проф. учеб. заведений / Н. Н. Глущенко, Т. В. Плетенева, В. А. Попков; Под ред. Т. В. Плетеневой. — М.: Издательский центр "Академия", 2004. — 384 с.
4. Драго Р. Физические методы в химии – М.: Мир, 1981
5. Кольтгоф И.М., Стенгер В.А. Объемный анализ В 2 томах – М.: Государственное научно-техническое издательство химической литературы, 1950
6. Коренман И.М. Фотометрический анализ – М.: Химия, 1970
7. Коростелев П. П, Фотометрический и комплексометрический анализ в металлургии – М.: Металлургия, 1984, 272 с.
8. Логинова Н. В., Полозов Г. И. Введение в фармацевтическую химию: Учеб. пособие – Мн.: БГУ, 2003.-250 с.
9. Мелентьева Г. А., Антонова Л. А. Фармацевтическая химия. — М.: Медицина, 1985.— 480 с.
10. Мискнджьян С.П. Кравченюк Л.П. Полярография лекарственных препаратов. – К.: Вища школа, 1976. 232 с
11. Фармацевтическая химия: Учеб. пособие / Под ред. Л.П.Арзамасцева. – М.: ГЭОТАР-МЕД, 2004. - 640 с.
12. Фармацевтический анализ лекарственных средств / Под общей редакцией В.А.Шаповаловой – Харьков: ИМП "Рубикон", 1995
13. Фармацевтичний аналіз: Навч. посіб. для студ. вищ. фармац. навч. закл. III—IV рівнів акредитації/П.О. Безуглий, В. О. Грудько, С. Г. Леонова та ін.; За ред. П.О. Безуглого,— X.: Вид-во НФАУ; Золоті сторінки, 2001.— 240 с.
14. Халецкий A.M. Фармацевтическая химия – Ленинград: Медицина, 1966
Похожие работы
.......................................................... 10 1. Введение Аргентометрические методы анализа относятся к методам осаждения, которые нашли мировое применение для анализа лекарственных средств. Аргентометрию используют для анализа роданидов, хлоридов, бромидов, йодидов щелочноземельных металлов и органических оснований. Рабочим раствором является раствор AgNO30,1М, а в ...

... сконто, дилерские. Заключение В ходе написания курсовой работы было проведено маркетинговое исследование группы нестероидных противовоспалительных средств с углублённым товароведческим анализом лекарственного препарата Мовалис, Определение положения препарата и фирмы-производителя на отечественном рынке. Данный препарат является высокоэффективным оригинальным продуктом немецкой фирмы Берингер ...
... как «варка», обжигание, охлаждение, промывание, «растирание» и смешение. Ему знакомы были химические превращения в виде брожения, свертывания, растворения, возгонки и другие. 2.3 Развитие технологии лекарственных средств в эпоху Возрождения Медицина фармация эпохи Возрождения характеризовались устремлением к опытному знанию. В борьбе за создание лекарств на основе точных химических знаний ...



еренных и тропических регионов всего мира, за исключением Австралии. Только в Америке естественно произрастает около 500 видов шалфеев. Шалфей лекарственный - Salvia officinalis Укр. назв. Шавлія лікарська Сем. Яснотковые - Lamiaceae Другие названия: шалфий, шальвия, шавлий, шалфей обыкновенный Ботаническая характеристика. Полукустарник высотой 20-50 см. В нижней части стебли ветвистые, ...
0 комментариев