Навигация
Таурин защищает клеточную мембрану, регулируя её состав и жёсткость
90483
знака
8
таблиц
12
изображений
4. Таурин защищает клеточную мембрану, регулируя её состав и жёсткость.
5. Таурин участвует в воспалительных ответах организма и, крайне необходим для защиты клеток крови. Он обладает детоксикационными свойствами, нейтрализует сильный окислитель гипохлорную кислоту, которая генерируется при окислительном взрыве нейтрофилов (как известно, эти клетки участвуют в иммунном ответе).
1.1.3 Названия препаратов с действующим веществом таурин* (тaurine*)
Дибикор (Dibicoram); Тауфон – АКОС (Tauranumx – AKOS); Тауфорин раствор 4%; Таурин (Taurin); Тауфона раствор 4% (глазные капли) (Solutio Таufoni 4%); Тауфон (Taufonum); Тауфона таблетки (Tabulettae Taufoni).
Препарат «Тауфон»
Латинское название: Taufonum (2-Аминоэтансульфоновая кислота).
Фармакологические группы: Белки и аминокислоты. Другие метаболики. Действующее вещество (международное непатентованное название) таурин* (тaurine*).
Фармакологические свойства: Тауфон относится к аминокислотным препаратам, стимулирующим репаративные и регенеративные процессы при заболеваниях сетчатки глаза дистрофического характера, травматических повреждениях тканей последнего, патологических процессах, сопровождающихся резким нарушением метаболизма этих тканей. Как серосодержащая аминокислота препарат способствует нормализации функций клеточных мембран, оптимизации энергетических и обменных процессов, поддержанию постоянства электролитного состава цитоплазмы клеток, торможению синоптической передачи.
Применение: Дистрофические поражения сетчатой оболочки глаза, в том числе наследственные тапеторетинальные дегенерации, дистрофии роговицы, катаракта, травмы роговицы, открытоугольная глаукома, сердечно-сосудистая недостаточность различной этиологии (в том числе на фоне интоксикации сердечными гликозидами). Также назначается при лечении органа зрения совместно с другими препаратами.
Противопоказания: Повышенная индивидуальная чувствительность к препарату.
Побочные действия: Аллергические реакции.
Форма выпуска: Для медицинского применения таурин выпускается в виде 4% водного раствора под названием «Тауфон» во флаконах из бесцветного стекла по 5 мл или 10 мл и в ампулах по 1 мл. Хранение: в прохладном, защищенном от света месте. 10 мл препарата содержат 0,4 г таурина. Этот препарат выбран нами для исследований, потому что отпускается без рецепта врача. Используется в виде бесцветного прозрачного стерильного раствора с рН 5-6,5.
1.2 Методы определения аминокислотПоскольку таурин часто рассматривают в ряду аминокислот, в нейтральной среде он существует в виде цвиттер-иона. Поэтому для его определения целесообразно рассмотреть методы анализа аминокислот.
В настоящее время разделение и определение аминокислот в различных биологических объектах является важной задачей клинической биохимии и аналитической химии. В связи с этим быстро развиваются методы анализа аминокислот и их смесей. К таким методам относятся: метод обращенно-фазовой ВЭЖХ с флуоресцентным или электрохимическим детектированием [6,7] (способы пред- и постколоночной дериватизации аминокислот), реакция с о-фталевым альдегидом в присутствии нуклеофильных агентов, которую широко используют для чувствительного электрохимического, спектрометрического и флуориметрическое анализа аминокислот [8], метод капиллярного электрофореза [9], метод алкалиметрического титрования [10,11] и др.
В ВЭЖХ аминокислоты определяют с использованием различных детекторов: УФ, лазерного флуориметрического (ЛИФ), электрохимического, а также рефрактометрического. Все методы детектирования, кроме последнего, требуют предварительной процедуры дериватизации. Для получения производных аминокислот с целью их последующего определения используют такие соединения, как о-фталевый альдегид, фенил-изотиоцианат (УФ-детектирование), флуоресцеин-изотиоцианат, флуоресцамин (ЛИФ-детектирование).
Серосодержащие аминокислоты обычно определяют методами хроматографии [12], потенциометрии, а также иодометрическим титрованием. Среди электрохимических методов анализа для определения серосодержащих аминокислот широко применяют вольтамперометрию [13,14,15].
Аминокислоты и пептиды можно легко обнаружить методом хроматографии на бумаге или в тонком слое целлюлозы, используя небольшие количества материала. Бумажная и тонкослойная хроматография чаще используются для качественного анализа. Для определения используются различные буферы и органические растворители, при использовании которых достигается разделение аминокислот при одномерном фракционировании. Не разделенные аминокислоты могут быть разделены в результате проведения хроматографии при других pH во втором (перпендикулярном) направлении. Среди реагентов дающих широкий диапазон цветов при взаимодействии с аминокислотами можно отметить: нингидрин, флуоресцамин, изатин. Аминогруппы могут реагировать со свободными альдегидными группами, содержащимися в бумаге. Образующиеся основания Шиффа дают синюю флуоресценцию в УФ-свете. Йод не вызывает разрушения аминокислот. Хлор пригоден для определения N–блокируемых аминокислот [2]. Быстро развивается лигандообменный хроматографический анализ аминокислот и пептидов на силикагельных сорбентах в присутствии ионов меди.
1.2.1 Метод кислотно-основного титрованияМетоды кислотно-основного титрования [10,11] основаны на использовании реакции между кислотами и основаниями. Алкалиметрическое титрование (щелочами) применяют для определения сильных и слабых кислот, кислых солей, солей слабых оснований и органических соединений, обладающих кислыми свойствами (кислоты, фенолы). Органические (слабые) кислоты способны оттитровываться раствором NaOH, причем образующийся анион кислоты вступает в реакцию гидролиза, и среда становится щелочной:
RCOOH + OH– D Н2O + RCOO–.
Таурин, вследствие амфотерного характера не удается непосредственно титровать раствором щелочи. Титрование оказывается возможным, если блокировать аминогруппу действием формальдегида:
H2N—(CH2)2—SO3OH + CH2O = CH2=N—(CH2)2—SO3H + Н2O.
Образующееся соединение можно титровать алкалиметрически с индикатором фенолфталеином в соответствии с уравнением реакции [5]:
CH2=N—(CH2)2—SO3H + NaOH D CH2=N—(CH2)2—SO3Na + Н2O.
1.2.2 Анализ аминокислот методом тонкослойной хроматографииРазработка методов количественного и качественного анализов аминокислот (АК) является важной задачей многих отраслей науки: медицины, биохимии, микробиологии, пищевой промышленности, фармакологии и сельского хозяйства. В настоящее время наиболее эффективно при массовых анализах может быть использована тонкослойная хроматография [12] (ТСХ). Денситометрия применяется для количественной тонкослойной хроматографии в качестве альтернативы методу высокоэффективной жидкостной хроматографии (ВЭЖХ), требующей дорогостоящего оборудования и расходных материалов. Применяют методы разделения свободных аминокислот и их производных с использованием одномерного и двухмерного вариантов тонкослойной хроматографии с последующей денситометрической обработкой хроматограмм. Программа "Dens" позволяет обрабатывать хроматограммы, полученные в двух вариантах с относительной погрешностью не более 3%. Двухмерная ТСХ дает возможность разделить большое количество соединений (до 22 наиболее важных аминокислот, в том числе и таурин) на коротком расстоянии проявления (стандартная пластина размером 10х10 см). Одномерная ТСХ позволяет анализировать до десяти и более образцов аминокислот одновременно. В работе [12] показана возможность разделения смеси 22-х свободных аминокислот методом двухмерной ТСХ (рис. 1.1).

Рис. 1.1. Вид хроматограммы при ТСХ – разделении аминокислот с использованием нингидрина как проявителя.
В последнее время широко используются AAA-Direct [17] системы для анализа аминокислот. Для такого анализа, характерна комбинация анионо-обменной хроматографии и амперометрического определения.
Достоинствами этого метода являются:
· Пределы определения от минимального до среднего диапазона (в пикомолях), без дериваций.
· Приблизительно в 50 раз более чувствительный, чем анализ на основе нингидрина, и конкурентоспособный с методом предколоночной деривации.
· Определяет аминокислоты, фосфоаминокислоты, углеводы, аминосахариды – за один отбор.
· Быстрый количественный анализ аминокислот, таких как триптофан и серосодержащие аминокислоты.
BioLC® AAA-Direct конфигурация анализа аминокислот – усовершенствованное определение аминокислот. В отличие от других существующих методик, аминокислоты определяются непосредственно. С высокочувствительной, встроенной функцией импульсного амперометрического определения нет необходимости в предварительной или постколоночной деривации. Такая функция предлагает прямое определение первичных и вторичных аминокислот, аминосахаридов, фосфоаминокислот и основных продуктов окисления серосодержащих аминокислот (напр. цистеиновая кислота) в фемтомолях – пикомолях пределах диапазона определения с чувствительностью в 50 раз выше, чем в методике, основанной на нингидрине.
1.2.3 Электрохимические методы определения аминонокислотМетод обращенно-фазовой ВЭЖХ с электрохимическим детектором [6] позволяет определить аминокислоты на уровне пикомоль. По этому методу можно определить 15 аминокислот в сыворотке крови человека. Так как немодифицированные аминокислоты не обладают электрохимической активностью, для детектирования их переводят в производные. Разделение производных проводят в градиентном или изократическом режимах элюирования. Из реагентов для получения производных только о-фталевый альдегид (ОФА), нафталин-2,3-дикарбоксиальдегид и 7-фтор-4-нитробензо-2-окса-1,3-диазол образуют с аминокислотами производные, обладающие электрохимической активностью. В качестве серосодержащих компонентов о-фталевого реагента используют 2-меркаптоэтанол или сульфит натрия. Общая продолжительность разделения 80 мин. Пределы обнаружения 0,5-5 пмоль. Методика применена для определения глутаминовой кислоты, аспарагина, серина, глутамина, гистидина, таурина, аланина, аргинина, метионина, изолейцина, орнитина, лейцина, фенилаланина, лизина и триптофана в сыворотке крови человека.
Для анализа нейромедиаторных аминокислот в реальных объектах используют обращено-фазовую ВЭЖХ с флуоресцентным или электрохимическим детектированием [7]. И поскольку, аминокислоты – нелетучие, неокрашенные соединения, слабо поглощающие в ультрафиолетовой области спектра, для их обнаружения также используется перевод в производные, обладающие флуоресцентной и электрохимической активностью. Наиболее широко применяется способ пред- и постколоночной дериватизации аминокислот, содержащих первичную аминогруппу, является образование изоиндолов при реакции с о-фталевым альдегидом и тиолами. Используя данную методику, можно в режиме изократического элюирования количественно определить глутаминовую кислоту, аспарагин, серин, глутамин, гистидин, таурин, аланин, аргинин и гаммааминомасляную кислоту в спинномозговой жидкости за 55 мин. Предел обнаружения 0,5-10 пмоль.
Реакцию о-фталевого альдегида с аминосодержащими соединениями в присутствии нуклеофильных агентов широко используют для чувствительного электрохимического, спектрометрического и флуориметрическое определения аминокислот [8]. В результате этой реакции образуются интенсивно флуоресцирующие продукты. С аминокислотами реакция идет по схеме:

где R-остаток аминокислоты, НХ-нуклеофильный агент, соединение Ι-замещенный изоиндол. В качестве нуклеофильных агентов могут выступать алкилмеркаптаны, меркаптопроизводные спиртов и органических кислот, а также сульфит- и цианид-ионы. Аналитические характеристики метода не уступают методу с использованием 2-меркаптоэтанола, а устойчивость аналитической формы – замещенных изоиндолов – существенно выше.
Разработан экспресс-метод идентификации и определения 11 аминокислот в их смеси с использованием прибора капиллярного электрофореза без их предварительной дериватизации и модифицирующих добавок к буферному раствору [9]. Содержание компонентов определяют с помощью фотометрического детектора. Этот метод в отличие от метода ВЭЖХ обладает рядом преимуществ: высокой эффективностью разделения, малым расходом реактивов, экспрессностью анализа и простотой аппаратурного оформления. Разделения смеси аминокислот в капилляре добиваются использованием различного рода добавок к фоновому электролиту. В частности, применяют метанол, ацетон, смесь раствора тетрабората натрия и изопропанола. Время анализа составляет ~15 мин. Диапазон определяемых концентраций 1-1000 мг/л. По методике можно определить глутаминовую кислоту, глутамин, аргинин, метионин, изолейцин, лейцин, фенилаланин, триптофан и др.
В последнее время широкое распространение в вольтамперометрии органических соединений получили химически модифицированные электроды (ХМЭ). Отличительной особенностью этих электродов является высокая селективность, которая достигается в результате взаимодействия модификатор-анализируемый компонент. Так, для вольтамперометрического определения цистеина используют угольно-пастовый электрод (УПЭ), модифицированный циклогексилбутиратом кобальта (II), меди (II), эфиром дибензо-18-краун-6 и его производными. Накопление аминокислоты на поверхности этих электродов происходит в виде соответствующего комплекса. Электрод, модифицированный оксидом рутения (IV), можно использовать для определения цистеина и цистина [14]. Способ инверсионно-вольтамперометрического определения позволяет анализировать такие серосодержащие аминокислоты, как цистеин, гомоцистеин и глутатион на УПЭ, модифицированных краун-эфирами дибензо-18-краун-6 или дибромдибензо-18-краун-6 [13]. Диапазон определяемых концентраций (2-5)·10-8 моль/л. В условиях проточно-инжекционного анализа разработана методика электрокаталитического определения серосодержащих аминокислот на графитовом электроде, модифицированном неорганической пленкой из гексацианоферрата (II) рутения (III) [15]. В качестве графитового материала используют стеклоуглерод или угольную пасту.
1.2.4 Фотометрические методыФотометрические методы основаны на измерении поглощения веществом светового излучения. В фотометрии применимы химические реакции, в результате которых получаются окрашенные продукты постоянного состава с высокой интенсивностью окраски. Фотометрические реакции органических соединений основаны на введении или создании в молекуле органического соединения системы сопряженных связей и образовании комплексных соединений. В фотометрических определениях аминокислот в качестве реагентов используют ароматические альдегиды (с образованием оснований Шиффа); ароматические амины (продукт – азосоединение); 1,2-нафтохинон-4-сульфокислоту (продукт – индонафтол); нингидрин (продукт – фиолетовый Руэмана) [18].
Определение тауфона в воздухе с 1,2-нафтохинон-4-сульфокислотой
Измерение концентрации таурина (в работе [21] – тауфона) в воздухе рабочей зоны используется как метод контроля на промышленных предприятиях. Метод основан на реакции взаимодействия тауфона с 1,2-нафтохинон-4-сульфокислотой в щелочной среде и последующем фотометрическом измерении окрашенного в желтый цвет продукта реакции при длине волны 440 нм. Отбор проб проводят концентрированием на фильтр. Нижний предел измерения содержания тауфона в анализируемом объеме раствора - 25 мкг. Нижний предел измерения концентрации тауфона в воздухе (при отборе 10 л воздуха) - 2,5 мг/м 3. Диапазон измеряемых концентраций в воздухе от 2,5 до 25 мг/м 3. Метод избирателен на стадии сушки и фасовки продукта. Определению тауфона мешают амины. Суммарная погрешность измерения не превышает ±15%. Время выполнения измерения, включая отбор проб - 40 мин.
Степень десорбции тауфона составляет 98,5%. Количественное определение содержания тауфона (мкг) в анализируемой пробе проводят по предварительно построенному градуировочному графику.
Определения, основанные на реакции с нингидрином
Растворы аминокислот, полипептидов, пептонов и первичных аминов при нагревании с нингидрином (1,2,3-индантрион) (НГ) приобретают синюю или фиолетовую окраску. Реакции между НГ и указанными соединениями протекают сложно (вещества претерпевают глубокое превращение). Предполагают, что сначала НГ восстанавливается, а аминокислоты окисляются, что сопровождается их декарбоксилированием и дезаминированием.
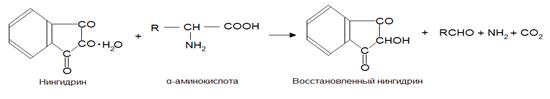
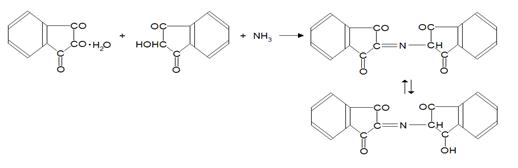
При дальнейшем взаимодействии избытка НГ с восстановленным НГ и аммиаком образуется окрашенный продукт конденсации.
Образующееся соединение имеет фиолетово-синюю окраску (λ max=570 нм). Нингидриновая реакция неспецифична, так как окрашенный продукт с нингидрином дают также NH3 и другие соединения, содержащие аминогруппу (в том числе белки и пептиды). Однако реакции с этими соединениями осуществляются без выделения СО2 (нингидриновая реакция с выделением СО2 специфична только для α-аминокислот). Реакцию используют для колориметрического количественного определения α-аминокислот, в том числе в автоматических аминокислотных анализаторах [18].
Конечным продуктом является фиолетовый (пурпурный) Руэмана. Казалось бы, что во всех случаях окраска раствора должна быть одинаковой. Однако возможны и другие процессы, как, например, взаимодействие образовавшихся альдегидов (побочные продукты) с аминокислотами, приводящие к появлению по-разному окрашенных продуктов [18].
В работе [19] предлагается другой механизм нингидриновой реакции:
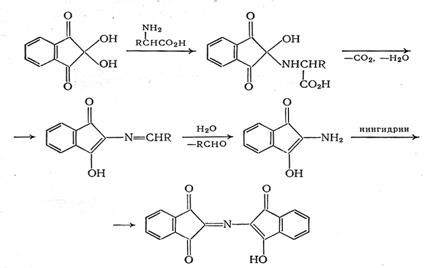
Выход продукта реакции зависит от свойств определяемой аминокислоты. Наиболее интенсивные окраски при прочих равных условиях наблюдаются при определении глицина, изолейцина, лейцина, норлейцина; несколько менее интенсивная окраска - при реакции с серином, фенилаланином, цистеином. Величины e510 [18] продуктов реакции лежат в пределах 1,8·104-3,3·104. Окрашенные продукты реакции нестабильны, и интенсивность окраски раствора довольно быстро уменьшается. Для стабилизации в состав реактива вводят хлорид кадмия, который с фиолетовым Руэмана образует устойчивые комплексные соединения. Присутствие кадмия влияет также на скорость реакции и на спектральную характеристику продукта. Хлорид кадмия можно заменить разбавленным раствором сульфата меди, который с фиолетовым Руэмана образует оранжево-красное соединение с lmax=530 нм.
Таким образом, значения молярного коэффициента поглощения продуктов реакции с нингидрином зависят от условий и в расчете на определяемое вещество, в зависимости от выхода реакции, составляют п·103 - 20·103.
В целом, для нингидриновой реакции характерна высокая чувствительность, поскольку отдельные ее стадии отличаются хорошими выходами и воспроизводимостью.
В работе [18] приведен также ряд методических рекомендаций для определения α-аминокислот, в том числе и таурина:
I. Смешивают 1 мл раствора, содержащего 0,02—0,4 мг α -аминокислоты, с 0,5 мл буферного раствора (рН 5,3—5,4) и 0,5 мл 3%-ного раствора нингидрина в метилцеллозольве. Нагревают 15 мин при 100°С, после чего добавляют 5мл 50%-ного изопропилового спирта и взбалтывают. После охлаждения до комнатной температуры красный раствор фотометрируют при 570 нм. Таким способом определяют аланин, аспарагиновую кислоту, аспарагин, валин, глицин, глутамин, глутаминовую кислоту, гистидин, изолейцин, лизин, орнитин, метионин, серии, таурин, треонин, тирозин, фенилаланин, этаноламин, а также аммиак. При определении пролина и оксипролина получают желтый раствор, который фотометрируют при 440 нм. Для приготовления буферного раствора растворяют 270 г ацетата натрия в 200 мл воды, добавляют 50 мл ледяной уксусной кислоты и воду до 750 мл. К 500 мл этого раствора добавляют 10 мл 0,01 М раствора NaCN.
II. Раствор в 80%-ном этиловом спирте, содержащий около 5 мкг аминокислоты, смешивают с 2 мл 0,2%-ного раствора нингидрина в изобутиловом спирте и этим же спиртом разбавляют до объема 10 мл. Нагревают 3 мин при 80ºС, затем охлаждают до 22ºС и фотометрируют при длинах волн от 530 до 560 нм. Так определяют аланин, аспарагин, валин, глицин, изолейцин, лизин, фенилаланин и ряд других аминокислот.
III. К 1 мл анализируемого раствора прибавляют 1 мл ледяной уксусной кислоты и 1 мл реактива, нагревают 1 ч при 100°С. После охлаждения разбавляют ледяной уксусной кислотой до объема 5 мл и через 1 ч красные растворы фотометрируют: в случае определения лизина при 500 нм; оксилизина - 450 нм; орнитина и пролина - 515 нм; цистеина - 570 нм. Для приготовления реактива при 70°С растворяют 0,25 г нингидрина в смеси 4 мл Н3РО4 (1 : 1) и 6 мл ледяной уксусной кислоты.
IV. Для анализа аминокислот в природных водах сначала проводят концентрирование. Пропускают 1 л исследуемой воды со скоростью 10—12 капель/мин через колонку (1 x 20 см) с катионитом КУ-2 в Н+-форме. Затем аминокислоты десорбируют с колонки 80 мл 2 н раствора NaOH. Полученный раствор выпаривают досуха при 100°С и остаток растворяют в 2 мл воды. Этот концентрат смешивают с 1 мл реактива, добавляют 2 капли насыщенного водного раствора CdCl2 и 5 мл н-бутилового спирта. Нагревают 15 мин при 100°С и после охлаждения добавляют 1 мл 25%-ного раствора сегнетовой соли. Слой бутилового спирта отделяют, разбавляют этим же спиртом до объема 6 мл и фотометрируют при 505 нм. Калибровочный график строят, используя стандартные растворы: аминоуксусной кислоты в (2—30 мкг в пробе в пересчете на азот). Отмечается, что интенсивность и устойчивость окраски в среде бутилового спирта выше, чем в воде. Реактив готовят растворением 75 мг СdCl2, 6 мл воды, 0,3 мл ледяной уксусной кислоты и 2 г нингидрина в 100 мл ацетона.
Для некоторых аминопроизводных (пролина, эфедрина) приводятся условия экстракционно-фотометрических определений [18].
Нингидриновая реакция использована в работе [20] для определения капролактама (КЛ) при концентрациях от 0,1 до 20,0 мг/л в сточных водах предприятий по его производству и переработке. В работе указано, что спектрофотометрический метод определения КЛ с нингидрином, несмотря на высокое значение эффективного молярного коэффициента светопоглощения e = 10600, имеет низкую чувствительность — 50 мг/л, что обусловлено пятидесятикратным разбавлением пробы в ходе анализа. При реакции с аминокислотой нингидрин восстанавливается до гидриндантина, а затем конденсируется с аммиаком и второй молекулой нингидрина, образуя краситель типа мурексида — дикетогидриндилидендикетогидриндамин (ДИДА). Аммиак образуется в результате окислительного расщепления аминокислот нингидрином. Реакция протекает только в присутствии вещества, способного восстановить нингидрин до гидриндантина. Поэтому предлагается в реагентную смесь кроме нингидрина вводить гидриндантин. Авторы отмечают, что вместо метилцеллозольва лучше использовать менее токсичный этилцеллозольв. Этилцеллозольв позволяет удержать продукты реакции в растворе без разбавления и повышает интенсивность окраски так, что значение e (М-1·см-1) увеличивается с 10600 до 19600, приближаясь к e ДИДА (21600). Область максимального светопоглощения окрашенными растворами находится в интервале длин волн от 560 до 585 нм. Так, оптическая плотность при 570 нм выше оптических плотностей при указанных длинах волн всего на 0,6%.
Предлагается [20] следующий раствор реагентов, свежеприготовленный: растворяют 2 г нингидрина и 0,3 г гидриндантина в 75 мл этилцеллозольва и добавляют 25 мл буферного раствора с pH = 6,5.
Для проведения нингидриновой реакции пипеткой отбирают 15 мл подготовленной для анализа сточной воды в пробирку, 8 мл раствора реагентов. Пробирку закрывают пробкой, перемешивают и на 22 мин помещают в баню с кипящей водой. Охлаждают до комнатной температуры водопроводной водой, и после выравнивания температур растворов в пробирках измеряют оптическую плотность по отношению к дистиллированной воде, в том числе и холостой опыт. Для приготовления буферного раствора с рН=6,5 в дистиллированной воде растворяют 544 г уксуснокислого натрия (гидрат) и 4 мл ледяной уксусной кислоты (плотность 1049 кг/м3) и доводят объем до 1 л. Для получения градуировочной зависимости готовят стандартные водные растворы КЛ с концентрацией 0,1; 0,5;...; 20,0 мг/л.
Из приведенных методик можно выделить факторы, которые необходимо учитывать и проверять при постановке (апробации) методики определения таурина:
· в качестве органического растворителя лучше использовать этилцеллозольв, который повышает интенсивность окраски раствора и менее токсичен, чем метилцеллозольв;
· уточнение рН буферного раствора, поскольку в литературных источниках приводятся границы 5,3 - 6,5;
· уточнение температуры реакции и режима нагрева;
· проверка необходимости введения гидриндантина в смесь реагентов;
· проверка встречающихся указаний на стабилизирующее действие спирта в составе реактива.
1.3 Мягкие контактные линзыКонтактные (т.е. надевание непосредственно на глазное яблоко под веки) линзы получили в последнее время большое распространение для улучшения зрения при близорукости, дальнозоркости, астигматизме, старческой дальнозоркости, а также для усиления или изменения цвета глаз. В разных странах ими пользуется от 2 до 10% населения. Первые контактные линзы созданы в начале 20-го века и были изготовлены из стекла, далее появились жесткие контактные линзы из полиметилметакрилата, в 60-е годы разработаны первые мягкие линзы из НЕМА, в 90-е – кислородопроницаемые жесткие линзы.
1.3.1 Основные характеристики мягких контактных линзМягкие контактные линзы (МКЛ) (рис. 1.2) [22] изготавливают из гидро-фильных полимеров, которые легко поглощают воду до определенной максимальной концентрации, уровень которой определяется такими физическими параметрами как температура, давление, рН и др.

Рис. 1.2. Мягкие контактные линзы и материалы для их изготовления.
Гидрогелем называется состояние полимерного каркаса с включенной в него водой.
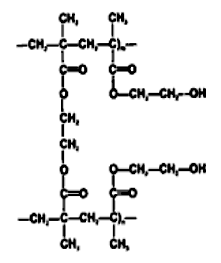
Рис. 2.3. Две цепочки гидроксиэтилметакрилата
Полимерный каркас может содержать различные гидрофильные группы и поперечные сшивки, которые и определяют равновесное состояние наполненного водой гидрогеля. Гидрофильными группами могут быть гидроксильные, амидные, лактамные и карбоксильные группы. Обычно используемым для сшивок агентом является этиленгликоль-диметакрилат (EGDMA). Без сшивок большинство гидрофильных полимеров растворилось бы в воде. Способность гидрогеля всасывать воду приводит к образованию водных каналов для передачи кислорода. Первые гидрогельные линзы были изготовлены чешским ученым Отто Вихтерле из гидрогеля рНЕМА (поли-2-гидроксиэтилметакрилат (рис. 2.3)); они оказались слишком толстыми и пропускали кислорода лишь ненамного больше, чем жесткие газонепроницаемые линзы из РММА (полиметилметакрилата). Революция в мире контактных линз произошла, когда стало возможным изготовление тонких линз с большой кислородопроницаемостью. Появление этих линз стимулировало поиски новых гидрогельных материалов, которые стали бы еще более физиологически совершенными.
Строение гидрогелей
Гидрогели представляют собой поперечно сшитые пористые, хорошо набухающие, но не растворяющиеся в воде полимеры. Обычно их получают полимеризацией водорастворимых ненасыщенных соединений в присутствии бифункционального сшивающего агента. В своем исходном состоянии до гидратации они похожи на жесткие полимеры - негибкие, ломкие и жесткие. При погружении в воду гидроксильные группы сухого полимера притягивают молекулы воды, и полимер поглощает воду. Объем поглощенной воды зависит от количества гидроксильных компонентов в его структуре. При насыщении водой полимер становится мягким и гибким.
Гидрогели имеют аморфное строение. Структура гидрогеля пронизана многочисленными порами, размеры и число которых у разных материалов сильно отличаются. Однако размеры пор (0,5-3,5 мкм) слишком малы для проникновения микроорганизмов, если структура полимера не повреждена. В то же время, многие ионы, консервирующие вещества и растворимые в воде препараты типа стероидов и антибиотиков могут с легкостью диффундировать как в гидрогель, так и в обратном направлении.
Основные характеристики МКЛ
Содержание воды в контактной линзе является одним из главных параметров МКЛ. Высокое содержание воды обеспечивает комфортность ношения линзы и снабжение роговицы кислородом. Вода обеспечивает продвижение кислорода через материал гидрогелевой линзы. Молекулы кислорода растворяются в воде и перемещаются через материал линзы к роговице.
Кислородная проницаемость критична для мягких контактных линз, так как «слезный насос» недостаточно эффективен для обеспечения роговицы кислородом. Большая часть необходимого роговице кислорода поступает сквозь линзу. Для характеристики кислородной проницаемости материала (но не конкретной линзы определенной толщины) используется коэффициент кислородной проницаемости (Dk). (Здесь D - коэффициент диффузии, k - коэффициент растворимости; в практике врача эти параметры по отдельности практически не встречаются).
Кислородная проницаемость материала прямо пропорциональна содержанию в нем воды и не зависит от толщины материала. Для характеристики способности конкретной линзы пропускать кислород используется коэффициент пропускания кислорода - Dk/L, где L - толщина линзы (обычно берется толщина линзы в центре). Этот коэффициент уже является характеристикой конкретной линзы и зависит, в частности, от ее толщины. Например, контактные линзы для коррекции сильно выраженной миопии, будучи очень тонкими в центральной зоне, позволяют кислороду легко проникать через них (Dk/L будет большим). С другой стороны, линзы для коррекции афакии очень толстые в центре и плохо пропускают кислород (Dk/L будет низким).
При снижении содержания воды происходит соответствующее снижение Dk/L. При этом могут изменяться и другие параметры линзы, что может повлиять на посадку линз. Снижение содержания воды на 20% приводит к снижению кислородной проницаемости приблизительно в 2 раза.
У большинства современных МКЛ кислородопроницаемость определяется в большей степени уровнем гидратации, чем природой полимерной структуры. Главным недостатком высокогидрофильных линз является их высокая чувствительность к механическим повреждениям, по сравнению с линзами со средним содержанием воды. Высокогидрофильные линзы, если сделать их слишком тонкими, могут даже вызывать повреждение эпителия роговицы, из-за его обезвоживания in situ.
Для изготовления более качественных МКЛ ведутся постоянные поиски новых материалов с более высоким содержанием воды, повышенной кислородной проницаемостью, увеличенной прочностью.
1.3.2 Применение МКЛКонтактные линзы в течение длительного времени служили, главным образом, средством оптической коррекции зрения. Линзы стали использоваться в лечении некоторых заболеваний глаза в качестве искусственной повязки для роговицы и средства введения лекарственных веществ в глаз. Однако если применение МКЛ с бандажной целью уже вошло в практику офтальмологов, то вопросы, связанные с введением лекарственных веществ в глаз с помощью линз, находится в стадии разработки. Известно, что МКЛ, пропитанные лекарственными веществами, продлевают их лечебное действие и вследствие этого являются более эффективным методом введения препаратов в орган зрения по сравнению с инстилляционным [23].
Для изготовления МКЛ применяются полимерные материалы на основе гидрогелей. Благодаря свойствам гидрогелей, обеспечивающим диффузию электролитов, кислорода и углекислого газа, мягкие линзы в меньшей степени, чем жесткие, влияют на метаболизм роговицы. Это дает возможность использовать их при заболеваниях роговицы с целью ее защиты. Сорбционно-десорбционные свойства гидрофильных материалов обуславливают применение линз, изготовленных из них, в качестве резервуара лекарственных препаратов, вводимых в глаз. Кислородная проницаемость и пропускаемость являются сложными процессами и в существенной степени зависят от содержания воды в материале, конструкции линз, температуры и типа мономера. Слезная пленка является основным поставщиком питательных веществ - кислорода, глюкозы, солей и минеральных веществ в роговицу. Кислород из воздуха содержится в слезной пленке в растворенном состоянии. Без контактных линз в открытые глаза может поступать до 21% всего кислорода воздуха. При закрытых глазах и без линз (во время сна) количество кислорода снижается до 7%. Контактные линзы значительно затрудняют проникновения кислорода в глаз. При закрытых глазах, например во время сна с контактными линзами для длительного ношения, процессы жизнедеятельности в роговице могут снизиться. Подбирая линзы, врачи обычно отдают предпочтение линзам, при ношении которых кислородоснабжение во время сна максимально. В целом, проницаемость линз для кислорода тем выше, чем больше воды они содержат и чем они тоньше. Все мягкие материалы для контактных линз за исключением новых линз, кремнийорганических компоненты, обладают способностью поглощать воду. В соответствии с долей содержащейся воды материалы для контактных линз разделяют на 3 категории: с низким содержанием воды - 35-45%; со средним содержанием воды - 45-60% и с высоким содержанием воды - 65-90% [22].
Применение МКЛ при лечении больных с различными повреждениями и заболеваниями глаз[24]:
· термические ожоги. Цель: снятие болевого синдрома, профилактика инфекционных осложнений;
· химические ожоги, комбинированные термомеханические поражения. Цель: снятие болевого синдрома, профилактика инфекционных осложнений;
· непротяженные раны роговицы с адаптированными краями. Цель: бандаж, профилактика инфекционных осложнений;
· протяженные и многолоскутные раны роговицы (после наложения узловых швов). Цель: устранение раздражения, вызванного узловыми швами, герметизация передней камеры, профилактика инфекционных осложнений;
· эпителиально-эндотелиальная дистрофия роговицы. Цель: перевод отечной стадии в сухую, снятие болевого синдрома.
В проблемной научно-исследовательской лаборатории КемГУ разработан высокогидрофильный полимерный материал для МКЛ «Кемерон-1», признанный соответствующим зарубежным аналогам и рекомендованный к промышленному производству [26]. «Кемерон-1» представляет собой сополимер на основе N-винилпирролидона и метилметакрилата, синтезированный в присутствии дивинилового сшивающего агента методом радиационной (g-излучение 60Со) блочной полимеризации.
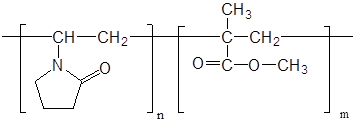
«Кемерон-1» наряду с хорошими оптическими свойствами (прозрачность, стабильность показателя преломления) обладает гибкостью, эластичностью, биологической инертностью и применяется для изготовления мягких контактных линз, используемых в бандажных целях. Исходный «Кемерон-1» – твердый стеклоподобный материал, после набухания становится эластичным и гибким.
Возможность применения МКЛ в качестве средства для введения лекарственных веществ в орган зрения зависит от их абсорбции (поглощения) материалом линзы и последующей десорбции [27,28]. Исследование этих процессов для МКЛ из «Кемерон-1» на примере глазных капель «Тауфон» (4% водный раствор таурина – 2-аминоэтансульфоновой кислоты) и входит в задачи дипломной работы.
2. ОБЪЕКТЫ И МЕТОДИКИ ЭКСПЕРИМЕНТА
2.1 Объект исследования
Объектом исследования в данной работе являются модельные калиброванные МКЛ из материала «Кемерон-1». В исходном состоянии до гидратации линзы представляют собой жесткие полимеры – негибкие и ломкие. При погружении в воду полимер по мере насыщения становится мягким и гибким. У набухшей линзы увеличивается ее масса и объем. Такая линза хранится в водном растворе.
В качестве вещества для исследования обменных свойств, был взят препарат «Тауфон» (разд. 2.1.5.), представляющий собой 4%-й водный раствор таурина – 2-аминоэтансульфоновой кислоты. Этот препарат выбран нами для исследования, поскольку отпускается без рецепта врача, довольно часто применяется для лечения органа зрения как совместно с другими лекарственными препаратами, так и отдельно, а литературные данные по сорбции и десорбции таурина материалом МКЛ отсутствуют.
2.2 Реактивы и аппаратура, используемые в работеХарактеристики исходных веществ:
1. Глазные капли «Тауфон» (производитель: ООО Славянская аптека, Р.№002492/01-2003; ЗАО фармацевтическая фирма «ЛЕККО», Р.№002696/01-2003).
2. Натрий уксуснокислый, 3- водный, ЧДА по ГОСТ 199-78.
3. Уксусная кислота ледяная по ГОСТ 61-75.
4. Олово двухлористое, 2- водное, ЧДА по ГОСТ 36-78.
5. Нингидрин (НГ), 1-водный, ЧДА ОКП 26 3812 0052, ТУ 6-09-10-1384-79.
6. Этилцеллозольв технический по ГОСТ 8313-88.
7. Спирт этиловый, чистый по ТУ 6-09-1710-77.
8. Формалин, 36,9% технический по ГОСТ 1625-89.
9. Гидроксид натрия, ЧДА по ГОСТ 4328-77.
10. Кислота соляная, фиксанал по ТУ 6-09-2540-72.
11. Натрия хлорид, раствор изотонический 0,9% (производитель: ОАО «БИОХИМИК», Р.№002134/01-2003).
12. Бидистиллированная вода.
Характеристики используемой аппаратуры:
1. Колбы вместимостью 100 см3, с пробками.
2. Колбы для титрования вместимостью 100 мл.
3. Бюретка, емкостью 25 мл.
4. Набор пробирок со стеклянными пробками.
5. Пипетки вместимостью 1, 2, 5 и 10 мл.
6. Водяная баня с ячейками для вертикальной установки колб в воду.
7. Фотоэлектроколориметр ФЭК-56М, снабженный кассетой с 9 светофильтрами [25].
8. Набор кювет с l = 1 см для фотометрирования.
2.3 Методики исследования 2.3.1 Методики определения тауринаМетодика кислотно-основного титрования
В связи с отсутствием стандартного вещества работа проводилась только с использованием готовых лекарственных форм капель (4% растворы таурина). Для использования капель в качестве рабочих растворов необходимо было независимым методом установить исходную концентрацию таурина в препарате. С этой целью удобен метод кислотно-основного титрования с предварительным блокированием аминогруппы формальдегидом [5]. Таким способом определяются только большие концентрации таурина, для более чувствительного анализа метод непригоден.
Концентрацию таурина [16] устанавливают следующим образом: 5 мл препарата помещают в коническую колбу вместимостью 100 мл, прибавляют 30 мл Н2O, 7 мл формалина и оставляют закрытую колбу на 10 мин. Затем титруют стандартизованным по 0,1000 M раствору HCl 0,1 М раствором NaOH до появления слабо-розовой окраски (индикатор-фенолфталеин, 0,06 мл). Параллельно проводят контрольный опыт.
1 мл 0,1 М раствора NaOH соответствует 0,01252 г C2H7NSO3 (таурина). Согласно инструкции к глазным каплям содержание таурина (в «Тауфоне») должно быть от 0,0380 до 0,0420 г в 1 мл препарата (4 %).
Концентрацию таурина рассчитывают по формуле:
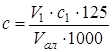 , [г/мл], (3.1)
, [г/мл], (3.1)
где V1 − объем NaOH, пошедший на титрование таурина, мл;
с1 – концентрация NaOH, моль/л;
Vал − объем препарата, мл;
125 – молярная масса таурина, г/моль.
Методика фотометрического определения
Для определения низких концентраций таурина был апробирован фотометрический метод определения α-аминокислот по реакции с нингидрином (НГ) с учетом рекомендаций работ [18-20] (разд. 2.2.4). Сущность его состоит в том, что растворы аминокислот при нагревании с нингидрином приобретают синюю или фиолетовую окраску. Фотометрические реакции очень чувствительны к чистоте реактивов, температуре и pH среды [18].
В работе изменены условия проведения фотометрической реакции (рН буферного раствора, состав смеси реагентов, режим нагревания), позволившие получить устойчивую окраску продукта с таурином, увеличить интенсивность окраски и уменьшить поглощение холостого опыта. Для проведения этой реакции важным является использование одного типа посуды (одинаковая толщина стекла).
Подготовка к анализу
1. Приготовление рабочего раствора:
В колбу объемом 100 мл вносят стандартизованный методом кислотно-основного титрования препарат «Тауфон» и доводят до метки бидистиллированной водой (раствор А, концентрация таурина в котором составляет 3,20·10-3 моль/л). В мерную колбу объемом 50 мл вносят 10 мл раствора А и доводят до метки бидистиллированной водой (раствор В с концентрацией таурина 6,40·10-4 моль/л).
2. Приготовление раствора реагентов:
Раствор реагентов, свежеприготовленный. Растворяют 2 г нингидрина в 37,5 мл этилцеллозольва и добавляют 37,5 мл этилового спирта.
3. Приготовление буферного раствора с рН 6,2. В дистиллированной воде растворяют 544 г уксуснокислого натрия (тригидрат) добавляют 4 мл ледяной уксусной кислоты (плотность 1049 кг/м3) и доводят объем до 1 л, затем доводят уксусной кислотой до нужного рН.
Ход анализа
В колбу на 100 мл пипеткой отбирают 3,00 мл раствора реагентов, 2,00 мл буферного раствора, 5,00 мл раствора, содержащего таурин (общий объем пробы 10,00 мл). При больших концентрациях таурина проводят промежуточное разбавление. После перемешивания помещают в баню с кипящей водой на 10 мин (колбы при нагревании открытые). Затем охлаждают до комнатной температуры водопроводной водой, и после выдерживания для выравнивания температур растворов в колбах измеряют оптическую плотность по отношению к дистиллированной воде при λ=570 нм. Параллельно проводят холостой опыт, в котором раствор таурина заменяют бидистиллированной водой [20]. За аналитический сигнал принимают разность полученных значений оптической плотности.
Для получения градуировочной зависимости в выбранных условиях готовят водные растворы таурина с концентрацией 0,06·10-4;……;1,0·10-4 моль/л соответствующим разбавлением раствора В.
Методика получения гидриндантина
Гидриндантин (ГД) получают восстановлением нингидрина. В 1 л дистиллированной воды растворяют при 90°С 40 г нингидрина и в 200 мл при 40°С 40 г аскорбиновой кислоты. Раствор кислоты при перемешивании вводят в раствор нингидрина, после перемешивания в течение 30 мин гидриндантин отфильтровывают с помощью водоструйного насоса на стеклянном фильтре, здесь же промывают дистиллированной водой и сушат в вакуумном эксикаторе над Р2О5.
Методика очистки этилцеллозольва
Технический этилцеллозольв очищают от содержащихся в нем перекисей путем кипячения в колбе с обратным холодильником с добавкой гидроксида натрия или хлористого олова (15-20 г на 0,5 л). Кипячение проводится в течение 15-20 минут в круглодонной колбе на воздушной бане. После этого колбу подсоединяют к водяному холодильнику с алонжем, который опускают в колбу-приемник. Перегонку ведут со скоростью 5-10 мл/мин. Собирают фракцию в интервале температур 135-136°С (около 350 мл). Перегнанный растворитель хранится в емкости из темного стекла в прохладном месте. Контроль содержания перекисей проводится периодически по реакции с иодидом калия.
2.3.2 Методика определения сорбции и десорбции тауринаОпределение сорбции и десорбции таурина МКЛ проводили с использованием фотометрической методики.
Исследование обменных свойств МКЛ проводили по схеме:
· измерение массы сухих линз;
· измерение массы насыщенных водой линз;
· определение методом кислотно-основного титрования концентрации исходного препарата;
· выдерживание МКЛ в препарате в течение определенного времени и определение концентрации таурина в растворе после извлечения линзы;
· выдерживание МКЛ, содержащих таурин, в воде или в физрастворе для десорбции и определение концентрации таурина в вытяжке при определенной частоте сменяемости воды и физраствора;
· расчет массы таурина в МКЛ по разности масс таурина в исходном препарате и в препарате после вымачивания линз;
· расчет сорбции и десорбции таурина МКЛ.
Методика проведения сорбции таурина МКЛ из материала «Кемерон-1»
Для изучения сорбции таурина линзы замачиваются в фиксированный объем препарата (2,00 мл) на определенное время. По истечении этого времени линзы извлекают из раствора таурина. Этот раствор используется для дальнейшего анализа. Для анализа раствора таурина после сорбции использовалась фотометрическая методика (разд. 3.3.1). Анализируемый раствор таурина разбавляется в 500 раз. При объеме пробы для анализа менее 5,00 мл для сохранения общего объема раствора при проведении реакции 10 мл в реакционную колбу добавлялось соответствующее количество воды.
Массу сорбированного вещества (Δm) находят по разности масс в исходном растворе капель (mисх) и после сорбции (mп.).
Массу таурина в каплях после сорбции рассчитывают по формуле:
mп = с · K · Vпробы · V1 · 125 · 10-3/ Vал, г (3.2)
где с − концентрация таурина в растворе, определяемая по уравнению градуировочной зависимости, моль/л;
Vпробы − объем анализируемого раствора, мл;
V1 − объем фотометрируемого раствора, мл;
Vал − объем аликвоты раствора таурина взятого на анализ, мл;
К − коэффициент разбавления;
125 – молярная масса таурина, г/моль.
Доля сорбированного вещества оценивается по отношению к массе сухой линзы (Δm/![]() безвод) и по отношению к mисх.
безвод) и по отношению к mисх.
Методика проведения десорбции таурина из МКЛ
Изучение десорбции таурина проводится следующим образом: после вымачивания линз в препарате для сорбции их извлекают из фиксированного объема раствора таурина и помещают в такой же фиксированный объем бидистиллированной воды или физраствора. Через определенные промежутки времени (30 мин.) линзы извлекают и снова помещают в такой же объем свежеприготовленной воды или физраствора (2,00 мл). Так повторяют несколько раз. Воду и физраствор после десорбции используют для анализа. Анализируемый раствор разбавляется в 50 раз. Для сравнения проводится десорбция однократно в суммарный объем.
Массу десорбированного вещества вычисляют по формуле (3.2).
2.4 Обработка результатов анализаУравнение линейного градуировочного графика получают методом наименьших квадратов (МНК) [29], позволяющим вычислить коэффициенты a и b в уравнении: y = a + bx.
b = (n Σxiyi – Σxi Σyi)/(n Σxi2 – (Σxi)2)
a = (Σyi – bΣxi)/n
Оценивают точность параметров a и b, для этого оценивают дисперсию S2yx экспериментальных точек:
S2yx · (n – 2) = Σyi2 – aΣyi – bΣxiyi
Дисперсию констант a и b вычисляют по уравнениям:
S2b = (n · S2yx)/(n Σxi2 – (Σxi)2)
S2a = (S2b / n) · Σxi2
Зная дисперсии констант a и b, можно рассчитать доверительные интервалы:
∆b = Sb · τα,ν
∆a = Sa · τα,ν
Окончательный вид уравнения прямой:
y = (a±∆a) + (b±∆b)x
Вычисление метрологических характеристик методики и результатов анализа:
xан = (y – a)/b,
Sx![]() =
=  .
.
Доверительный интервал результата анализа:
∆xан = Sxан · τα,ν.
Предел обнаружения (xmin):
![]() ; xmin = сmin =
; xmin = сmin = ![]() .
.
При проведении параллельных измерений проводилась статистическая обработка результатов по стандартной методике [29] с вычислением границ доверительного интервала (С) и относительного стандартного отклонения Sr.
3. Результаты эксперимента и их обсуждение 3.1 Изучение условий проведения фотометрической реакции таурина с нингидрином
В разделе 2.3.1 дано описание методики фотометрического определения таурина в условиях проведения нингидриновой реакции с образованием фиолетового (синего) Руэмана [18-20], установленных в работе после предварительно проведенных исследований. Были апробированы некоторые рекомендации для определения капролактама [20] и аминокислот [18].
Поиск условий проведения фотометрической реакции с нингидрином с целью определения низких концентраций таурина заключался в следующем:
· изучение влияния рН буферного раствора;
· изучение влияния температуры и режима нагревания;
· изучение влияния добавок гидриндантина (ГД), органических растворителей;
· изучение влияния порядка введения реактивов на величину устойчивого аналитического сигнала.
Попытка получения окрашенного в сине-фиолетовый цвет продукта реакции нингидрина (НГ) с таурином по методике [18] (разд. 2.2.4 – пропись I) не удалась. Причиной этого может быть, с одной стороны, неполное соблюдение условий, связанных с невозможностью использования NaCN в составе буферного раствора, и отсутствие рекомендованного изопропанола. С другой стороны, что вероятнее, указанное значение рН буфера (5,3-5,4) не подходит для проведения реакции (образуется продукт желто-оранжевого цвета).
Поэтому возникла необходимость поиска других условий, в частности, с использованием рекомендаций работы [20]. При проведении эксперимента варьировались: соотношение компонентов, порядок их введения, рН буферного раствора, концентрация таурина. Измерение оптической плотности окрашенного продукта проводилось при 570 нм. Результаты представлены в таблице 3.1.
Таблица 3.1.
Влияние условий проведения реакции таурина с нингидрином на получение окрашенного продукта
| № | Условия опыта | стаурина, моль/л | Результаты (А570) |
| ||||
| 11. | 4 мл раствора таурина; 2 мл раствора реагентов Ι; 3 мл этилцеллозольва; 1 мл буферного раствора рН 6 | 6,4·10-4 | Окраска отсутствует. |
| ||||
| 22. | 4 мл раствора таурина; 1 мл буферного раствора рН 6; 2 мл раствора реагентов Ι; после охлаждения пробы 3 мл этилцеллозольва | 6,4·10-4 | Окраска отсутствует. |
| ||||
| 33. | 1 мл раствора таурина; 2 мл раствора реагентов Ι; 3 мл этилцеллозольва; 1 мл буферного раствора рН 6,55 | 6,4·10-4 | Окраска отсутствует. |
| ||||
| 44. | 2 мл раствора реагентов Ι; 1 мл буферного раствора рН 6; 1 мл раствора таурина; 3 мл Н2O; 3 мл этилцеллозольва | 3,2·10-3 | Окраска отсутствует. |
| ||||
| 55. | 2 мл раствора реагентов Ι; 1 мл буферного раствора рН 6,55; 1 мл раствора таурина; 3 мл Н2O; 3 мл этилцеллозольва | 3,2·10-3 | Окраска отсутствует. |
| ||||
| 66. | 3 мл раствора реагентов ΙΙ; 2,5 мл буферного раствора рН 6,55; 2 мл раствора таурина; 2,5 мл Н2O (окрашенный продукт был разбавлен в 3 раза) | 3,2·10-3 | Продукт фиолетового цвета (0,972); окраска нестабильна. |
| ||||
| 77. | 3 мл раствора реагентов ΙΙ; 2,5 мл фосфатного буферного раствора рН 6,86; 2 мл раствора таурина; 2,5 мл Н2O | 3,2·10-3 | Окраска отсутствует. |
| ||||
| 88. | 3 мл раствора реагентов ΙΙ (готовился без ГД); 2,5 мл буферного раствора рН 6,55; 2 мл раствора таурина; 2,5 мл Н2O (окрашенный продукт был разбавлен в 3 раза) | 3,2·10-3 | Продукт фиолетового цвета (0,718); окраска нестабильна. |
| ||||
| 99. | 2,6 мл раствора реагентов ΙΙ; 0,9 мл буферного раствора рН 6,55; 0,5 мл раствора таурина; 6 мл Н2O | 3,2·10-3 | Окраска не стабильна (0,125) |
| ||||
| 110. | 2,6 мл раствора реагентов ΙΙ (готовился с избытком ГД (в 2 раза)); 0,9 мл буферного раствора рН 6,55; 0,5 мл раствора таурина; 6 мл Н2O | 3,2·10-3 | Окраска стабильна в течение 15 мин. (0,224) |
| ||||
| 111. | 3,5 мл раствора реагентов ΙΙ (готовился с избытком ГД (в 2 раза)); 0,9 мл буферного раствора рН 6,55; 0,5 мл раствора таурина; 5,1 мл Н2O | 3,2·10-3 | Окраска стабильна в течение 15 мин. (0,206) |
| ||||
| 112. | 2,6 мл раствора реагентов ΙΙ (готовился с избытком ГД (в 2 раза)); 1,5 мл буферного раствора рН 6,55; 0,5 мл раствора таурина; 5,4 мл Н2O | 3,2·10-3 | Окраска стабильна и более интенсивна (0,443) | ||||
| 113. | 3 мл раствора реагентов ΙΙ (готовился с избытком ГД (в 2 раза)); 2 мл буферного раствора рН 6,55; 0,5 мл раствора таурина; 4,5 мл Н2O | 3,2·10-3 | Окраска стабильна до 20 мин. и более интенсивна (0,684) | ||||
| 114. | 3 мл раствора реагентов ΙΙ (готовился с избытком ГД (в 2 раза)); 2 мл буферного раствора рН 6,55; 5 мл раствора таурина. Нагревание в закрытой колбе 20 мин. Градуировочная зависимость в интервале концентраций от 0,1·10-4 моль/л до 2,5·10-4 моль/л: y = (0,054 | 6,4·10-4 | Окраска стабильна до 20 мин. Раствор интенсивно окрашен. (1,573) | ||||
Примечания к таблице: Раствор реагентов Ι: 0,2 г НГ и 0,03 г ГД растворяли в 10 мл этилцеллозольва. Раствор реагентов ΙΙ по методике [20]: 0,2 г НГ и 0,03 г ГД растворяли в 7,5 мл этилцеллозольва.
При проведении фотометрической реакции необходимо было получить устойчивый аналитический сигнал в течение времени, достаточного для анализа, высокий и постоянный выход окрашенного продукта, обеспечивающий определение концентрации таурина менее 1·10-5 моль/л (для исследования десорбции). Наиболее подходящими условиями для получения красителя оказались результаты опытов с 2- кратным избытком ГД по отношению к рекомендуемому в методике [20] и увеличение количества буферного раствора практически в 2 раза (п.14 табл.4.1). В этом случае аналитический сигнал получается и при низких концентрациях таурина, а оптическая плотность стабильна в течение 20 мин (рис. 4.1). Для получения более стабильной окраски рекомендуют [18] проводить реакцию в присутствии солей меди или кадмия. В данном эксперименте в качестве стабилизатора использовался CuSO4 с концентрацией 10-3 моль/л (1 мл). Однако на устойчивость окраски добавка соли меди практически не повлияла, с течением времени она также убывает.
Таким образом, показано, что ГД увеличивает интенсивность окраски продукта реакции, повышает ее стабильность (рис. 3.1).
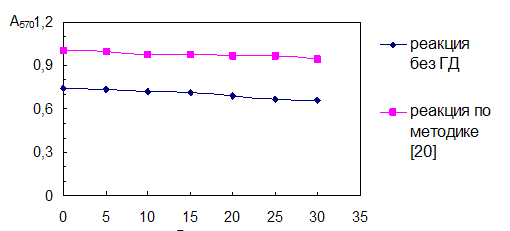
Рис. 3.1. Влияние ГД на получение и стабильность красителя
Однако введение ГД приводит к появлению плохо воспроизводимой окраски холостого опыта красных оттенков, которая налагается на окраску продукта, как это можно видеть из спектров поглощения (рис. 3.2а).
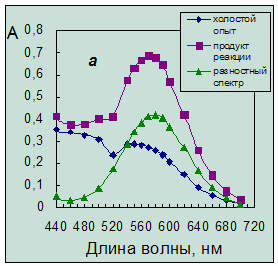
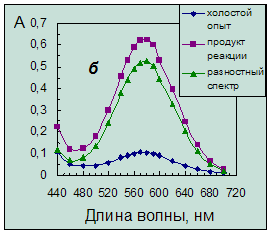
Рис. 3.2. Спектры поглощения продукта нингидриновой реакции.
Концентрация таурина 6,4·10-5 моль/л. Условия реакции:
а – в присутствии гидриндантина по п.14 (табл. 4.1) (разбавление в 3 раза);
б – без гидриндантина при рН 6,2 с добавкой этанола.
Анализируя полученные результаты, приведенные в табл. 3.1, можно сделать следующие обобщения:
1. Раствор реагентов Ι - “не работает”, т.к. все реакции с этой смесью не дали окрашенного продукта.
2. ГД повышает устойчивость окраски во времени; без ГД интенсивность окраски раствора ниже и устойчивость снижается.
3. Важна также последовательность добавления веществ для получения окрашенного продукта и дальнейшего анализа, а именно: раствор реагентов, буферный раствор, раствор таурина.
4. Необходимо использовать ацетатный буферный раствор с рН 6,50-6,55, в отличие от 5,3-5,4 по методике [18].
Таким образом, методика анализа с введением ГД имеет следующие недостатки: плохую воспроизводимость результатов, высокий аналитический сигнал холостого опыта (рис. 4.2а) и ограниченную стабильность продукта. В связи с этим был необходим поиск иных условий проведения реакции таурина с нингидрином без введения ГД.
Исследование влияния рН в интервале 5,1 – 6,5, создаваемом ацетатным буфером, показало, что синий продукт образуется при рН 6,5 и без ГД, в отличие от капролактама [20]. То есть для проведения нингидриновой реакции с таурином необходимы более высокие значения рН, чем указанные в работе [18]. При этом время нагревания при температуре 80ºС пришлось увеличить до 60 минут, вместо 22 мин [20].
Согласно литературным данным на стабильность и интенсивность окраски продукта оказывают влияние добавки спиртов в реакционную смесь. Вводить спирт для стабилизации окраски рекомендуется или после реакции (изопропиловый, амиловый) или во время реакции (этиловый вместе с изобутиловым) [18]. Введение в смесь реагентов этилового спирта и проведение реакции при температуре 100ºС в течение 10 минут позволило получить окрашенный продукт сине-фиолетового цвета (рис. 3.2б), устойчивый в течение суток. Порядок введения реактивов не влияет на аналитический сигнал. При этом аналитический сигнал холостого опыта существенно ниже, чем в присутствии ГД (рис. 3.2).
При использовании такого смешанного растворителя установлено, что наибольшая величина полезного аналитического сигнала получается при рН 6,2 и приготовлении раствора нингидрина в этилцеллозольве с добавлением спирта в соотношении 1:1. При рН 6,55 в присутствии этанола практически в 2 раза увеличивается оптическая плотность холостого опыта, что нежелательно. На образование окрашенного продукта влияет режим нагревания. Для лучшей повторяемости результатов в присутствии этанола, как показали наблюдения, нагревание следует проводить в вертикально установленных на кипящую водяную баню открытых колбах одинакового калибра. При этом получается окрашенный продукт, сходимость измерений оптической плотности которого (А570) на уровне 0,1 (при концентрации таурина 1,3·10-5 моль/л) при п = 6¸7 характеризуется Sr не превышающим 3%.
Таким образом, использование спирта позволило в определенной степени решить поставленные задачи. Установлены наиболее приемлемые условия проведения нингидриновой реакции с таурином: растворитель нингидрина – этилцеллозольв, стабилизатор окраски – этиловый спирт (их соотношение в смеси реагентов 1:1); буферный раствор с рН 6,2; температура реакции 100ºС; время нагревания 10 минут. Нагревание следует проводить в тонкостенных открытых колбах с большим свободным объемом (100 мл при объеме реакционной смеси 10 мл). Методические детали выполнения анализа в отработанных условиях приведены в разд. 2.3.1.
3.2 Характеристики градуировочной зависимости для определения таурина в выбранных условияхВ выбранных условиях проведения фотометрической реакции (разд. 2.3.1) была получена градуировочная зависимость оптической плотности растворов при 570 нм (А570) от концентрации таурина, линейная в диапазоне концентраций таурина 0,06·10-4 – 1,0·10-4 моль/л и проходящая через начало координат (рис. 3.3).
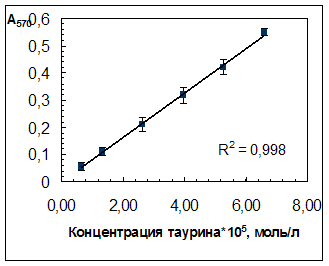
Рис. 4.3. Зависимость оптической плотности продукта нингидриновой реакции от концентрации таурина в растворе.
Исходная концентрация таурина в препарате «Тауфон», установленная методом кислотно-основного титрования, составила (0,0414 ± 0,0001) г/мл.
Характеристики градуировочной зависимости:
b = 8170; a = -0,0023 (не значим);
Sb = 174; Sа = 0,007;
Cb = 480;
y = (8170 ![]() 480)x;
480)x;
ymin = 0,020 (Аmin при 570 нм);
Предел обнаружения: сmin= 4,20·10-6 М (5,25·10-4 мг/мл).
Следует отметить, что характеристики градуировочной зависимости практически не изменяются в присутствии физраствора.
Значение экспериментального молярного коэффициента поглощения, близкое к 8100 М-1·см-1, занимает промежуточное среди указанных в работе [18,20] для продуктов с различными аминопроизводными (от 1,6·103-20·103). Это может быть связано с недостаточно высоким выходом продукта реакции, но стабильность и воспроизводимость окраски полученного в условиях методики красителя свидетельствует о ее применимости для проведения анализа.
Таблица 3.2.
Метрологические характеристики методики определения таурина с нингидрином в установленных условиях (р = 0,95).
| Введено, мкг | Найдено | |||
|
| d, % | Sr, % | ||
| 7,6 | п = 7 | 7,7 ± 0,2 | 1,3 | 2,9 |
| 16,5 | п = 5 | 17,0 ± 1,0 | 3,0 | 4,9 |
| 28,0 | п = 7 | 27,6 ± 1,0 | 1,4 | 1,5 |
| 66,1 | п = 5 | 65,9 ± 1,4 | 0,3 | 1,7 |
Примечание: 1) исходная концентрация таурина, установленная методом кислотно-основного титрования (разд. 3.3.1) 41,3 г/л (0,331 моль/л), разбавлена в 500 раз; взятые объемы, соответственно, равны 0,10; 0,20; 0,34 и 0,80 мл.
Отработанная методика имеет хорошие метрологические характеристики. Полученная градуировочная зависимость использовалась при исследовании обменных свойств МКЛ по отношению к препарату «Тауфон».
3.3 Результаты исследования обменных свойств МКЛ и их обсуждениеПодготовка линз к проведению исследования.
МКЛ на основе материала «Кемерон-1» были выточены специально для изучения обменных свойств и представляли собой в сухом состоянии прозрачные неэластичные образцы диаметром 12,00±0,01 мм и толщиной в центральной части 0,60 мм.
После взвешивания сухих МКЛ проводили их насыщение водой до постоянной массы. При набухании линзы увеличивались в размере и приобретали эластичность. Набухшие линзы хорошо встряхивали, а затем взвешивали (работа проводилась с 3 линзами). Взвешивание повторяли не менее 5 раз. Данные представлены в виде таблицы 3.2.
Таблица 3.3.
Массы линз из материала «Кемерон-1»
| № линзы |
(безводный полимер) | тл, г (гидратированный полимер) |
(гидратированный полимер) | С (гидратированный полимер) |
| 1 | 0,1082 | 0,3208; 0,3209; 0,3225; 0,3230; 0,3247 | 0,322 | 0,001 |
| 2 | 0,1069 | 0,3195; 0,3160; 0,3171; 0,3153; 0,3162 | 0,317 | 0,001 |
| 3 | 0,1068 | 0,3105; 0,3112; 0,3145; 0,3114; 0,3170 | 0,313 | 0,001 |
Сорбцию таурина МКЛ изучали следующим образом: каждую линзу (предварительно насыщенная до постоянной массы водой) помещали в 2 мл препарата на разное время до полного насыщения веществом. По истечении этого времени линзы извлекали, а оставшийся раствор анализировали в соответствии с отработанной методикой, отбирая на анализ определенную аликвоту раствора. Предварительная проверка показала, что независимо от объема аликвоты, определяемые значения количества таурина в растворе после извлечения таурина одной и той же линзой практически одинаковы (табл. 3.4), что подтверждает работоспособность методики (такая проверка тождественна проверке на отсутствие систематической погрешности путем кратного увеличения размера пробы).
Таблица 3.4.
Результаты анализа раствора после сорбции таурина одной линзой из объема раствора 2,00 мл с массой таурина 86,6 мг
| А570 | Vал, мл | с·105, моль/л | тп, мг | (mп ± С), мг |
| 0,095 0,095 0,095 0,095 0,095 0,100 0,095 | 0,2 | 1,12 | 70,0 | 70,7 ± 2,0 |
| 1,12 | 70,0 | |||
| 1,12 | 70,0 | |||
| 1,12 | 70,0 | |||
| 1,12 | 70,0 | |||
| 1,20 | 75,0 | |||
| 1,12 | 70,0 | |||
| 0,355 0,355 0,355 0,360 0,350 0,360 0,355 | 0,8 | 4,30 | 67,0 | 67,0 ± 2,0 |
| 4,30 | 67,0 | |||
| 4,30 | 67,0 | |||
| 4,36 | 68,0 | |||
| 4,23 | 66,0 | |||
| 4,36 | 68,0 | |||
| 4,30 | 67,0 |
Изучение динамики сорбции проводилось на трех линзах, характеристики которых представлены в табл. 3.1. Для трехчасовой сорбции проведена статистическая обработка результатов (табл. 1, Приложение 1). Относительное стандартное отклонение без поправки на массу линз не превышает 5% при определении таурина, оставшегося в растворе (тп) и 8% при определении массы поглощенного таурина (Dт).
Результаты сорбции в статических условиях при разном времени насыщения МКЛ в растворе «Тауфона» представлены в табл. 4.5 и на рис. 3.4.
Таблица 3.5.
Результаты сорбции таурина МКЛ из объема раствора 2,00 мл с массой таурина 86,6 мг
| t, ч | Масса в растворе после сорбции (mп), мг | Масса извлеченного одной линзой (Δm), мг | Степень сорбции (Δm / мг/г | Степень извлечения (Δm/ mисх), % |
| 0,5 | 78,7 | 7,9 | 74 | 9,1 |
| 1 | 76,0 | 10,6 | 99 | 12,2 |
| 2 | 72,5 | 14,1 | 132 | 16,3 |
| 3 | 71,5 | 17,9 | 168 | 20,7 |
| 4 | 70,2 | 16,4 | 154 | 18,9 |
| 24 | 71,3 | 15,3 | 143 | 17,7 |
 Рис. 4.4. Кривые сорбции таурина из раствора «Тауфона»
Рис. 4.4. Кривые сорбции таурина из раствора «Тауфона»
МКЛ из материала «Кемерон-1»
Анализируя полученные данные, можно сделать вывод о том, что полное насыщение препаратом линз происходит за три часа (~ 15-17 мг на одну линзу). Причем за первый час извлекается более 60%, а через 3 часа достигается сорбционная емкость. Ее величина составляет около 160 мг/г (0,16 г на 1 г безводного материала). Данных по сорбции таурина МКЛ нами не найдено. Можно, однако сравнить полученную величину сорбционной емкости с приведенными в работе [28] значениями при сорбции другими материалами МКЛ аминогликозидов и цефалоспоринов из глазных капель. В зависимости от химической природы сополимеров, влагосодержания гидрогелей и исходной концентрации капель она составляет 2 – 500 мг/г. На примере сорбции гентамицин-сульфата сополимером ГЭМА-ММА показано, что при уменьшении концентрации препарата в 10 раз сорбционная емкость уменьшается также практически в 10 раз: от 22 до 2 мг/г [28].
Если провести оценку коэффициента распределения таурина между гидрогелем и раствором в состоянии равновесия согласно [30] как отношение 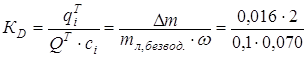 , то его величина составит 4,6. Его величина ближе к коэффициентам распределения, приведенным в работе [31] для гидрофильных веществ, поглощаемых в пленках гидрогелей из поли-2- оксиэтилметакрилата, сшитого этиленгликольдиметакрилатом (водопоглощение около 40%). Наибольшее значение (5,84) указано для метотрексата натрия. Для гидрофобных транспортируемых веществ KD имеют в десятки раз большие значения. Авторы считают, что в гидрогеле гидрофильные вещества диффундируют и распределяются, в основном, в псевдообъемной воде.
, то его величина составит 4,6. Его величина ближе к коэффициентам распределения, приведенным в работе [31] для гидрофильных веществ, поглощаемых в пленках гидрогелей из поли-2- оксиэтилметакрилата, сшитого этиленгликольдиметакрилатом (водопоглощение около 40%). Наибольшее значение (5,84) указано для метотрексата натрия. Для гидрофобных транспортируемых веществ KD имеют в десятки раз большие значения. Авторы считают, что в гидрогеле гидрофильные вещества диффундируют и распределяются, в основном, в псевдообъемной воде.
Согласно полученным результатам для насыщения МКЛ из материала «Кемерон-1» препаратом «Тауфон» достаточно трех часов.
3.3.2 Результаты изучения десорбции тауринаИсследование десорбции проводилось на тех же линзах, характеристики которых представлены в табл. 3.1.
При изучении десорбции линзу после насыщения препаратом извлекали из раствора «Тауфона» и помещали в склянки с 2 мл бидистиллированной воды или физраствора последовательно, выдерживая линзу в каждой из склянок по 30 минут. Такой схемой пытались смоделировать смену слезной жидкости. Полученные растворы анализировали. Общее время десорбции составило 1,5 часа. Выдерживание в воде или в физрастворе (замену склянок) прекращали, когда при анализе проб значения оптической плотности соответствовали холостому опыту. Для сравнения десорбция из предельно насыщенного таурином гидрогеля на основе «Кемерон-1» осуществлялась также в суммарный объем в течение 1,5 часов. Оценка погрешности определения массы десорбированного таурина показала, что относительное стандартное отклонение не превышает 3,5%, причем десорбция в воду и физраствор происходит одинаково (табл. 2, Приложение 1).
Результаты представлены в табл. 3.6 и на рис. 3.5.
Таблица 3.6.
Результаты исследования десорбции таурина линзами после выдерживания в бидистиллированной воде
| Сорбция | Десорбция | |||||
| Время сорбции, час | Масса таурина извлеченного одной линзой (Δm ± С), мг | Время десорб-ции, мин. | Vал, мл | с·105, моль/л | Масса десорбир. таурина mв, мг | Степень десорбции (mв/Δm), % |
| 0,5 | 7,9±2,0 | 30 | 0,5 | 1,62 | 4,0 | 50,6 |
| 30 | 1 | 0,06 | 0,1 | 1,3 | ||
| 30 | 3 | 0,00 | 0,0 | – | ||
| S mв = 4,1 | 51,9 | |||||
| 1 | 10,6±1,0 | 30 | 0,5 | 2,20 | 5,5 | 51,9 |
| 30 | 1 | 1,04 | 1,3 | 12,3 | ||
| 30 | 3 | 0,65 | 0,3 | 2,8 | ||
| S mв = 7,1 | 67,0 | |||||
| 2 | 14,1±1,0 | 30 | 0,5 | 2,12 | 5,0 | 35,5 |
| 30 | 1 | 0,88 | 1,1 | 7,8 | ||
| 30 | 3 | 0,50 | 0,1 | 0,7 | ||
| S mв = 6,2 | 44,0 | |||||
| 3 | 17,9±2,0 | 90 | 1 | 3,10 | 11,6 | 64,8 |
| 4 | 16,4±2,0 | 30 | 0,5 | 1,98 | 4,9 | 29,9 |
| 30 | 1 | 1,03 | 1,3 | 7,9 | ||
| 30 | 3 | 0,59 | 0,2 | 1,2 | ||
| S mв = 6,4 | 39,0 | |||||
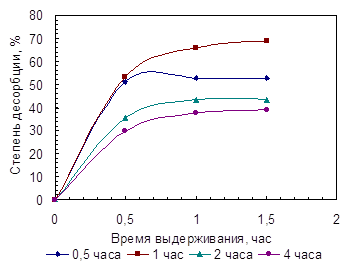
Рис. 3.5. Кривые десорбции таурина из МКЛ при указанном времени предварительной сорбции.
Полученные результаты показали, что степень десорбции таурина из МКЛ зависит от времени предварительного насыщения: при полуторачасовой десорбции она уменьшается от 52-67 до 39% в зависимости от времени сорбции (табл. 3.6, рис. 3.5). Причем, при малых временах насыщения (30 минут, 1 час) процесс десорбции идет быстрее (~50% за первые 0,5 часа) и полнее, чем при более длительном насыщении (за 2 и 4 часа – 30 и 35%, соответственно). Увеличение общего объема воды для десорбции таурина из насыщенной линзы приводит к возрастанию степени десорбции за те же 1,5 часа (табл. 3.6).
Таким образом, в данных условиях для таурина имеет место необратимая сорбция. Причиной подобного поведения может быть различие в размещении молекул таурина в объеме гидрогеля. При малых временах насыщения молекулы таурина при абсорбции диффундируют в псевдообъемной (не связанной) воде гидрогеля. Возможно также, что при этом задействован не весь объем гидрогеля. При больших временах насыщения могут иметь место более сложные процессы, обусловленные более глубоким проникновением молекул таурина в объем линзы. Возможна диффузия препарата в пограничную и связанную воду [31] гидрогеля, даже с частичным ее вытеснением вплоть до проявления специфических взаимодействий между функциональными группами полимера и молекулами таурина.
Удаление оставшегося в линзе таурина осуществляется нагреванием в склянке с водой на кипящей водяной бане в течение 20 минут с последующей заменой воды и выдерживания при комнатной температуре до восстановления массы линзы. Повторное использование линзы для исследований приводит к тем же результатам по сорбции (Приложение 1).
Если рассматривать гидрогель МКЛ как транспортное средство для доставки таурина в глаз, то можно провести простые расчеты. При инстилляционном введении препарата вводится 1,7 мг/капля или 3,3 мг/2 капли. Часть препарата сразу вымывается слезой. При постепенном поступлении таурина из насыщенной линзы может быть десорбировано ~6,5 мг.
Проведенное исследование не позволяет судить о скорости поступления препарата в реальных условиях. На основании выводов работы [27] можно предположить, что она на 10-15% ниже наблюдаемой.
Таким образом, полученные результаты свидетельствуют о возможности пролонгированного введения препарата в ткани глаза с использованием МКЛ из материала «Кемерон-1» в качестве транспортного средства.
ВЫВОДЫ
1. Выбраны условия проведения фотометрической реакции таурина с нингидрином: растворитель нингидрина – этилцеллозольв, стабилизатор окраски – этиловый спирт (их соотношение в смеси реагентов 1:1); буферный раствор с рН 6,2; температура реакции 100ºС; время нагревания 10 минут.
2. Получена градуировочная зависимость для определения таурина и определены ее характеристики: y = (8170 ![]() 480)x; ymin = 0,020; xmin = сmin = 4,20·10-6 М. Диапазон определяемых концентраций 6,5·10-6 – 7,0·10-5 моль/л.
480)x; ymin = 0,020; xmin = сmin = 4,20·10-6 М. Диапазон определяемых концентраций 6,5·10-6 – 7,0·10-5 моль/л.
3. Установлено, что время насыщения МКЛ таурином составляет 3 часа; масса таурина, поглощенного одной линзой, составляет 15 – 17 мг.
4. Показано, что степень десорбции таурина уменьшается с увеличением степени сорбции. Для насыщенных линз она достигает ~40%, причем более 50% поглощенного таурина десорбируется за первые 30 минут.
5. Показано, что МКЛ из материала «Кемерон-1» можно использовать для местного введения препарата «Тауфон» в орган зрения.
ЛИТЕРАТУРА
1. Википедия – свободная энциклопедия: статья таурин [Электронный ресурс] – Электрон. текстовая прогр. - http://ru.wikipedia.org.
2. Досон, Р. Справочник биохимика [Текст] / Р. Досон, Д. Эллиот, У. Элиот, К. Джонс. - М.: Мир. - 1991. – С. 32, 389.
3. Нефедов, Л.И. Таурин [Электронный ресурс] – Электрон. текстовая прогр. / Л.И. Нефедов, И.Д. Волотовский. // Национальная академия наук Беларуси институт биохимии. - http://bodrost.com.ua/taurin2.doc.
4. Пескова, Л.Н. Синтез таурина [Электронный ресурс] – Электрон. текстовая прогр. - http://www.wikiznanie.ru.
5. Гауптман, З. Органическая химия [Текст] / З. Гауптман, Ю. Трефе, Х. Ремане. - Leipzig. – С. 506.
6. Краснова, И.Н. Определение аминокислот в сыворотке крови человека методом обращенно-фазовой высокоэффективной жидкостной хроматографии в режиме изократического элюирования [Текст] / И.Н. Краснова, Л.А. Карцова, Ю.В. Черкас. // Журнал аналитической химии. - 2000. – Т. 55. - №1.
7. Анализ нейромедиаторных аминокислот и биогенных аминов в спинномозговой жидкости методом обращенно-фазовой высокоэффективной жидкостной хроматографии [Текст] / И.Н. Краснова, И.В. Колмакова, Л.А. Карцова. // Журнал аналитической химии. - 1997. – Т. 52. - №7.
8. Бекетов, В.И. Спектрофотометрическое и флуориметрическое определение аминокислот по реакции с о-фталевым альдегидом в присутствии сульфит- и цианид-ионов [Текст] / В.И. Бекетов, Р.Д. Воронина, Д.Г. Филатова, Н.Б. Зоров. // Журнал аналитической химии. - 2000. – Т. 55. - №12.
9. Манаенко, О.В. Экспресс-определение аминокислот методом капиллярного электрофореза без их предварительной дериватизации [Текст] / О.В. Манаенко, А.И. Сидоров, Э.М. Сульман. // Журнал аналитической химии. - 2003. – Т. 58. - №10.
10. Пономарев, В.Д. Количественный анализ [Текст]: аналитическая химия: в 2 т. / В.Д. Пономарев. - М.: Высшая школа, - 1982. – Т. 2 - С. 44.
11. Коренман, Я.И. Титриметрические методы анализа [Текст]: практикум по аналитической химии / Я.И. Коренман. – Воронеж: изд. Воронежского университета. - 1986. – 334 с.
12. Анализ аминокислот в тонкослойной хроматографии [Электронный ресурс] – Электрон. текстовая прогр. / Научно-производственный центр «ЛЕНХРОМ» (Патент РФ №2095808 "Способ разделения и детектирования аминокислот") - http://lenchrom.spb.ru.
13. Шайдарова, Л.Г. Инверсионно-вольтамперометрическое определение некоторых аминокислот на модифицированных краун-эфирами угольно-пастовых электродах [Текст] / Л.Г. Шайдарова, И.Л. Федорова, Н.А. Улахович, Г.К. Будников. // Журнал аналитической химии. - 1997. – Т. 52. - №3.
14. Шайдарова, Л.Г. Электрокаталитическое окисление цистеина и цистина на угольно-пастовом электроде модифицированном оксидом рутения (IV) [Текст] / Л.Г. Шайдарова, С.А. Зиганшина, Г.К. Будников. // Журнал аналитической химии. - 2003. – Т. 58. - №6.
15. Шайдарова, Л.Г. Электрокаталитическое окисление и проточно-инжекционное определение серосодержащих аминокислотна графитовых электродах, модифицированных пленкой из гексацианоферрата рутения [Текст] / Л.Г. Шайдарова, С.А. Зиганшина, Л.Н. Тихонова, Г.К. Будников // Журнал аналитической химии. - 2003. – Т. 58. - №12.
16. Раствор Тауфона 4% (глазные капли) [Текст] / Фармакопейная статья ФС 42-2852-97. – 3 с.
17. Биохроматография [Электронный ресурс] – Электрон. текстовая прогр. - http://envitec.com.ua/h/bioh.html.
18. Коренман, И.М. Фотометрический анализ: методы определения органических соединений [Текст] / И.М. Коренман. – М.: Химия, – 1970.–С. 166–170.
19. Травель, В.Ф. Органическая химия [Текст]: учебник для вузов: в 2 т. / В.Ф. Травель. – М.:ИКЦ «Академкнига». – 2004. – Т. 2– С. 514.
20. Чеджемов, Г.Х. Определение капролактама в сточных водах с нингидрином [Текст] / Г.Х. Чеджемов, Г.А. Шлепанова. // Химические волокна. – 1985. – №1. – С. 57.
21. Методические указания (разработанные Новокузнецким НИХФИ) [Текст] / Утверждено: и.о. Председателем Госкомсанэпиднадзора России - заместителем Главного государственного санитарного врача Российской Федерации Г.Г. Онищенко. 8 июня 1996 г // МУК 4.1.0.322-96 - http://www.vsestroi.ru.
22. Минаев, Ю.Л. Основные характеристики МКЛ [Электронный ресурс] – Электрон. текстовая прогр. / Ю.Л. Минаев // Приложение к журналу «Глаз» - М. - http://www.contlenses.com.
23. Применение МКЛ, насыщенных лекарственными препаратами, в лечении заболевания органа зрения [Текст] / Методические рекомендации: Министерство здравоохранения СССР. МНИИ глазных болезней им. Гельмгольца. - М. – 1987.
24. Ушаков, Н.А. О применении МКЛ при повреждении и заболевании глаз [Текст] / Н.А. Ушаков, Ю.П. Гудаковский, Э.В. Муравьева // Военно-медицинский журнал. – 1992. – №8.
25. Инструкция по эксплуатации фотоэлектроколориметра ФЭК-56М. - 27 с.
26. Пак, В.Х. Российский материал для мягких контактных линз [Текст] / В.Х. Пак, В.Д. Жевняк, Т.В. Дикунова, Ю.Ф. Хатминский, Е.В. Прозорова, // Глаз. – 2007. – №1.
27. Рыбакова, Е.Г. Закономерности десорбции лекарственных препаратов из мягких контактных линз [Текст]: сообщение 2: исследования in vivo / Е.Г. Рыбакова, С.Э. Аветисов, Г.А. Бадун, А.В. Краснянский. // Вестник офтальмологии. – 1996. – №1.
28. Даниличев, В.Ф. Лечебные мягкие контактные линзы на основе полимерных гидрогелей [Текст] / В.Ф. Даниличев, С.С. Иванчев, Н.А. Ушаков, В.Н. Павлюченко, В.А. Рейтузов, А.С. Бобашева, Э.В. Муравьева. // Глаз. – 2006. – № 5.
29. Дерффель, К. Статистика в аналитической химии [Текст] / К. Дерффель. – М.: Мир, – 1994. – 268 с.
30. Москвин, Л.Н. Методы разделения и концентрирования в аналитической химии [Текст] / Л.Н. Москвин, Л.Г. Царицына. – Л.: Химия, – 1991. – 256 с.
31. Роуленд, С. Вода в полимерах [Текст] / Пер. с англ. Под ред. С. Роуленд. – М.: Мир, – 1984. – С. 335-346.
Приложение 1
Таблица 1.
Статистические данные по сорбции таурина за 3 часа тремя МКЛ из раствора «Тауфона» объемом 2,00 мл с массой таурина 86,6 мг
| № МКЛ | А570 | Vал, мл | с·105, моль/л | тп, мг |
| Δm, мг |
| 1. | 0,355 0,355 0,355 0,360 0,350 0,360 0,355 | 0,8 | 4,30 | 67,0 | 67,0 ± 0,5 | 19,5 |
| 4,30 | 67,0 | |||||
| 4,30 | 67,0 | |||||
| 4,36 | 68,0 | |||||
| 4,23 | 66,0 | |||||
| 4,36 | 68,0 | |||||
| 4,30 | 67,0 | |||||
| 2. | 0,365 0,370 0,370 0,370 0,370 0,370 0,360 | 0,8 | 4,40 | 68,7 | 69,0 ± 1,0 | 17,1 |
| 4,48 | 70,0 | |||||
| 4,48 | 70,0 | |||||
| 4,48 | 70,0 | |||||
| 4,48 | 70,0 | |||||
| 4,48 | 70,0 | |||||
| 4,36 | 68,0 | |||||
| 3. | 0,375 0,375 0,370 0,365 0,365 0,365 0,370 | 0,8 | 4,50 | 70,0 | 69,0 ± 0,7 | 17,2 |
| 4,50 | 70,0 | |||||
| 4,48 | 70,0 | |||||
| 4,40 | 68,7 | |||||
| 4,40 | 68,7 | |||||
| 4,40 | 68,7 | |||||
| 4,48 | 70,0 |
Результаты по 3 линзам:
масса таурина, оставшегося в растворе: (![]() )= 68,3±2,9; S =1,3; Sr= 4,4%;
)= 68,3±2,9; S =1,3; Sr= 4,4%;
масса извлеченного (Dт) таурина: (![]() ± С) = 17,9 ± 3,2; S = 1,3; Sr = 7,3 %.
± С) = 17,9 ± 3,2; S = 1,3; Sr = 7,3 %.
Таблица 2.
Статистические данные по десорбции таурина из насыщенных МКЛ в воду и физраствор
| № МКЛ | А570 | Vал, мл | с·105, моль/л | mв, мг | (mв ±С), мг |
| 1. | 0,260 | 0,1 | 3,13 | 11,7 | 12,0 ± 0,1 |
| 0,265 | 3,19 | 12,0 | |||
| 0,265 | 3,19 | 12,0 | |||
| 0,270 | 3,25 | 12,0 | |||
| 0,265 | 3,19 | 12,0 | |||
| 0,270 | 3,25 | 12,0 | |||
| 2. | 0,260 | 0,1 | 3,13 | 11,7 | 11,7 ± 0,2 |
| 0,255 | 3,07 | 11,5 | |||
| 0,260 | 3,13 | 11,7 | |||
| 0,260 | 3,13 | 11,7 | |||
| 0,265 | 3,19 | 12,0 | |||
| 0,260 | 3,13 | 11,7 | |||
| 3. | 0,250 | 0,1 | 3,01 | 11,3 | 11,2 ± 0,2 |
| 0,245 | 2,90 | 11,0 | |||
| 0,245 | 2,90 | 11,0 | |||
| 0,250 | 3,01 | 11,3 | |||
| 0,250 | 3,01 | 11,3 | |||
| 0,255 | 3,07 | 11,5 |
Средние значения по трем линзам для массы десорбированного (тв) таурина:
(![]() ± С) = 11,6 ± 1,0; S = 0,41; Sr = 3,5 %.
± С) = 11,6 ± 1,0; S = 0,41; Sr = 3,5 %.
0 комментариев