Навигация
Роль наследственности и среды в наследственной патологии человека
79253
знака
0
таблиц
0
изображений
КАРАГАНДИНСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
Рефератна тему: «Роль наследственности и среды в наследственной патологии человека»
Составили: студентки ОМФ-123
Сайбель Наталия
Щеголева Светлана
Проверил: Бритько В.В.
КАРАГАНДА 2009
Содержание
1 Наследственные болезнии их виды
2 Моногенные и полигенные заболевания
3 Общие принципы лечения наследственных заболеваний.
4 Виды профилактики наследственных заболеваний.
5 Значение наследственной предрасположенности
6 Характеристики генных болезней
7 Критерии разных типов наследования
1 Наследственные болезни и их классификация
Наследственные болезни— заболевания человека, обусловленные хромосомными и генными мутациями. Нередко ошибочно термины «наследственная болезнь» и «врожденная болезнь» употребляются как синонимы, однако врожденными болезнями называют те заболевания, которые имеются уже при рождении ребенка и могут быть обусловлены как наследственными, так и экзогенными факторами. Таковы, например, пороки развития, связанные с воздействием на эмбрион и плод ионизирующего излучения, химических соединений, лекарственных средств, принимаемых матерью, а также внутриутробных инфекций.
Однако далеко не все наследственные болезни относят к врожденным, поскольку многие из них проявляются уже после периода новорожденности (например, хорея Гентингтона клинически обнаруживается после 40лет).
Наследственные и врожденные болезни являются причиной госпитализации детей почти в 30% случаев и даже больше (с учетом болезней неизвестной природы, которые в значительной степени могут быть вызваны генетическими факторами).
В качестве синонима термина «наследственные болезни» не следует также рассматривать термин «семейные болезни», т.к.семейные заболевания могут быть обусловлены не только наследственными факторами, но и условиями жизни, национальными либо профессиональными традициями семьи.
В зависимости от соотношения роли наследственных и экзогенных факторов в этиологии и патогенезе различных заболеваний все болезни человека условно можно разделить на три группы.
Первая группа— собственно наследственные болезни, т.е. болезни, при которых проявление патологической мутации как этиологического фактора практически не зависит от влияния окружающей среды, которая в этом случае определяет лишь степень выраженности симптомов болезни. К болезням первой группы относятся все хромосомные и генные наследственных болезней с полным проявлением, например болезнь Дауна, фенилкетонурия, гемофилия и др.
К болезням второй группы относят так называемые мультифакториальные болезни, в основе которых лежит взаимодействие генетических и средовых факторов. К болезням этой группы относятся гипертоническая болезнь, атеросклероз, язвенная болезнь желудка и двенадцатиперстной кишки, сахарный диабет, аллергические заболевания, многие пороки развития, определенные формы ожирения.
Генетические факторы, представленные определенной полигенной системой, обусловливают генетическую предрасположенность, которая может быть реализована при воздействии неблагоприятных или вредных факторов окружающей среды (физического или умственного переутомления, нарушения режима и сбалансированности питания ит.п.). Для одних из них влияние окружающей среды имеет большее, для других— меньшее значение.
К мультифакториальным болезням относят также состояния, при которых роль генетического фактора может играть один единственный мутантный ген, но проявляется это состояние также только при определенных условиях. Примером такого состояния может служить дефицит глюкозо-6-фосфат- дегидрогеназы.
Болезни третьей группы связаны исключительно с воздействием неблагоприятных или вредных факторов окружающей среды, наследственность в их возникновении практически не играет никакой роли. К этой группе относят травмы, ожоги, острые инфекционные болезни. Однако генетические факторы могут оказывать определенное влияние на течение патологического процесса, т. е. на темпы выздоровления, переход острых процессов в хронические, развитие декомпенсации функций пораженных органов. Наследственные болезни обычно подразделяют на три основные группы: моногенные, полигенные (мультифакториальные, или болезни с наследственным предрасположением) и хромосомные.
Клиническая классификация наследственных болезней построена по органному и системному принципам. Согласно этой классификации, выделяют наследственные болезни, нервной, эндокринной, дыхательной и сердечнососудистой систем, печени, желудочно-кишечного тракта, почек, системы крови, кожи, уха, носа, глаз и др. Такая классификация в значительной степени условна, т.к.большинство наследственных болезней. характеризуется вовлечением в патологический процесс нескольких органов или системным поражением тканей.
Моногенные болезни по типу наследования могут быть аутосомно-доминантными, аутосомно-рецессивными и сцепленными с полом; по фенотипическому проявлению— ферментопатиями (болезнями обмена веществ, в т.ч. болезнями, обусловленными нарушением репарации ДНК): болезнями, обусловленными молекулярной патологией структурных белков; иммунопатологией, в т.ч. болезнями, вызванными нарушениями в системе комплемента; нарушениями синтеза транспортных белков (в т.ч. белков крови) и пептидных гормонов; патологией свертывающей системы крови; дефектами механизма переноса веществ через клеточные мембраны. Среди моногенных болезней также выделяют группу синдромов с множественными врожденными пороками развития, при которых неуточнен первичный дефект мутантного гена. Моногенные болезни наследуются в полном соответствии с законами Менделя. Большинство известных наследственных болезней. обусловлено мутациями структурных генов, возможность этиологической роли мутаций генов-регуляторов при некоторых заболеваниях до сих пор доказана лишь косвенно.
Аутосомно-доминантный тип наследования характерен для наследственных болезней, в основе которых лежит нарушение синтеза структурных белков или белков, выполняющих специфические функции (например, гемоглобина). При аутосомно-доминантном типе наследования действие мутантного гена проявляется практически всегда. Больные мальчики и девочки рождаются с одинаковой частотой. Вероятность развития болезни в потомстве составляет 50%. Вновь возникшая мутация в гамете одного из родителей может привести к спорадическому случаю доминантной патологии. По аутосомно-доминантному типу наследуются Марфана синдром, болезнь Олбрайта, дизостозы, отосклероз, пароксизмальная миоплегия, талассемия и др.
При аутосомно-рецессивном типе наследования мутантный ген проявляется только в гомозиготном состоянии. Больные мальчики и девочки рождаются с одинаковой частотой. Вероятность рождения больного ребенка составляет 25%. Родители больных детей фенотипически могут быть здоровы, но являются гетерозиготными носителями мутантного гена. Аутосомно-рецессивный тип наследования более характерен для заболеваний, при которых нарушена функция одного или нескольких ферментов,— так называемый ферментопатий.
Рецессивное наследование, сцепленное с X-хромосомой, заключается в том, что действие мутантного гена проявляется только при XY-наборе половых хромосом, т.е. у мальчиков. Вероятность рождения больного мальчика у матери— носительницы мутантного гена— составляет 50%. Девочки практически здоровы, но половина из них является носительницами мутантного гена (так называемые кондукторами). Часто болезнь обнаруживается у сыновей сестер пробанда (человека, по отношению к которому составляется генеалогическое древо) или его двоюродных братьев по материнской линии. Больной отец не передает болезнь сыновьям. Этот тип наследования характерен для прогрессирующей мышечной дистрофии типа Дюшенна, гемофилии А и В, синдрома Леша— Найхана, болезни Гунтера, болезни Фабри, некоторых форм генетически обусловленной недостаточности глюкозо-6-фосфат— дегидрогеназы.
Доминантное наследование, сцепленное с X-хромосомой, заключается в том, что действие доминантного мутантного гена проявляется в любом наборе половых хромосом (XX, XY, ХО и др.). Заболевание более тяжело протекает у мальчиков. Среди детей больного мужчины в случае такого типа наследования все сыновья здоровы, все дочери поражены. Больные женщины передают измененный ген половине сыновей и дочерей. Этот тип наследования прослеживается, например, при фосфат-диабете.
Ферментопатии, относящиеся к моногенным наследственным болезням по фенотипическому проявлению, составляют наиболее обширную и лучше всего изученную группу наследственных болезней. Первичный дефект фермента расшифрован более чем для 200ферментопатии. Возможны следующие причины ферментопатии:
а) фермент не синтезируется совсем;
б) в молекуле фермента нарушена последовательность аминокислот, т.е. изменена его первичная структура;
в) отсутствует или неправильно синтезируется кофермент соответствующего фермента;
г) активность фермента изменена в связи с аномалиями в других ферментных системах;
д) блокада фермента обусловлена генетически детерминированным синтезом веществ, вызывающих инактивацию этого фермента.
Мутация гена может повлечь за собой нарушение синтеза белков, выполняющих пластические (структурные) функции. Нарушение синтеза структурных белков— вероятная причина таких заболеваний, как остеодисплазии и остеогенез несовершенный. Есть данные об определенной роли этих нарушений в патогенезе наследственных нефритоподобных заболеваний— синдрома Альпорта и семейной гематурии. Дисплазия ткани в результате аномалий в структуре белков может наблюдаться не только в почках, но и в любых других органах. Патология структурных белков характерна для большинства наследственных болезней, наследуемых по аутосомно-доминантному типу.
Мутация гена может привести к развитию болезней, вызванных иммунодефицитными состояниям. Наиболее тяжело протекает агаммаглобулинемия, особенно в сочетании с аплазией вилочковой железы. Причиной появления гемоглобина с аномальной структурой при серповидно-клеточной анемии является замена в его молекуле остатка глутаминовой кислоты на остаток валина. Подобная замена является результатом генной мутации. Это открытие послужило началом интенсивного изучения большой группы наследственной болезни, названных гемоглобинопатиями.
Известен ряд мутантных генов, контролирующих синтез факторов свертывания крови. Генетически детерминированные нарушения синтеза антигемофилического глобулина (фактор VIII) приводят к развитию гемофилииА. При нарушении синтеза тромбопластического компонента (фактор IX) развивается гемофилияВ. Недостаток предшественника тромбопластина лежит в основе патогенеза гемофилииС.
Генные мутации могут быть причиной нарушения механизма транспорта различных соединений через клеточные мембраны. Наиболее изучены наследственная патология транспорта аминокислот в кишечнике и почках, синдром мальабсорбции глюкозы и галактозы, отмечены генетически обусловленные нарушения нормального функционирования так называемый К+. Na+-иoннoro насоса (АТФ-азы) клетки. Известны заболевания, причиной которых служит дефект механизмов, ответственных за поддержание нормального градиента концентраций ионов К+ и Mg2+ по обе стороны клеточной мембраны, что клинически проявляется периодическими приступами тетании. Примером заболевания, вызванного генетически обусловленным дефектом механизма транспорта аминокислот через клеточные мембраны, является цистинурия, клиническим выражением которой является нефролитиаз и симптомы пиелонефрита. Классическая цистинурия обусловлена нарушением переноса ряда диаминокарбоновых кислот (аргинина, лизина) и цистина через клеточные мембраны, как в кишечнике, так и в почках. Патология реабсорбции глюкозы в почечных канальцах— почечная глюкозурия— связана с нарушением функции мембранных белков-переносчиков или с дефектами в системе обеспечения энергией процессов активного транспорта глюкозы; наследуется по аутосомно-доминантному типу. Нарушение реабсорбции бикарбонатов в проксимальных отделах почечных канальцев или нарушение секреции водородных ионов клетками почечного эпителия дистальных отделов почечных канальцев лежит в основе развития двух типов почечного канальцевого ацидоза.
Частота встречаемости моногенных наследственных болезней колеблется у разных этнических групп в разных географических зонах. В странах Западной Европы и СССР наиболее распространенными Н.б. являются галактоземия (1:20000— 1:40000), гистидинемия (1: 17000), фенилкетонурия (1:12000— 1:15000), цистинурия (1:14000), муковисцидоз (1:1200— 1:5000). Частота встречаемости гиперлипопротеинемий (в т.ч. и полигенно наследуемых форм) достигает 1:100— 1:200. К часто встречающимся наследственным болезням относятся гемофилия (1:10000; болеют мальчики), гипотиреоз (1:7000), синдром мальабсорбции (1:3000), адреногенитальный синдром (1:5000— 1:11000). Частота встречаемости значительного числа Н.б. обмена пока не установлена, хотя это не свидетельствует об их редкости.
Полигенные (мультифакториальные) наследственные болезни, или болезни с наследственным предрасположением, обусловлены взаимодействием нескольких (или многих) генов в полигенных системах и факторов окружающей среды. Патогенез болезней с наследственным предрасположением, несмотря на их распространенность, изучен недостаточно.
Большое значение имеет поиск фенотипических маркеров наследственной предрасположенности к определенному заболеванию, например, аллергический диатез может быть диагностирован на основании повышенного содержания в крови иммуноглобулинаЕ и повышенной экскреции с мочой минорных метаболитов триптофана. Определены биохимические маркеры наследственной предрасположенности к сахарному диабету (повышенная толерантность к глюкозе, содержание в крови иммунореактивного инсулина), конституционально-экзогенному ожирению, гипертонической болезни (гиперлипопротеинемия). Достигнуты успехи в изучении взаимосвязи между группами крови АВО, антигенами системы гистосовместимости HLA и возможностью развития некоторых болезней. Установлено, что для лиц с тканевым гаплотипомHLA В8 высок риск заболевания хроническим гепатитом, целиакией и миастенией; для лиц с гаплотипомHLA А2— хроническим гломерулонефритом, лейкозом; для лиц с гаплотипомHLA DW4— ревматоидным артритом, для лиц с гаплотипомHLA А1— атопической аллергией. Корреляция с системой HLA обнаружена примерно для 90заболеваний человека, многие из которых характеризуются нарушениями системы иммунитета.
Болезни с наследственным предрасположением также имеют особенности распространения в разных странах. Так, по данным Шандса (A.R.Shands), частота расщепления губы и нёба в Англии составляет 1:515, в Японии— 1:333, а врожденный вывих бедра в Японии наблюдается в 10раз чаще, чем в Англии.
Хромосомные болезни подразделяют на аномалии, обусловленные изменениями количества хромосом (полиплоидии, анеуплоидии) или структурными перестройками хромосом— делециями, инверсиями, транслокациями, дупликациями. Хромосомные мутации, возникшие в зародышевых клетках (гаметах), проявляются в так называемых полных формах. Нерасхождение хромосом и структурные изменения, появившиеся на ранних стадиях дробления зиготы, ведут к развитию мозаицизма. Вероятность появления хромосомных аберраций резко увеличивается в гаметах женщин старше 35лет.
Частота встречаемости всех хромосомных болезней среди новорожденных, по данным Кэбека (М.М.Kaback), составляет 5,6:1000, при этом все виды анеуплоидий, включая мозаичные формы, составляют 3,7:1000, трисомии по аутосомам и структурные перестройки— 1,9:1000. Половину всех случаев структурных перестроек хромосом представляют семейные случаи, все трисомии являются спорадическими случаями, т. е. следствием вновь возникших мутаций. По данным Полани (P.Polani), около 7% всех беременностей осложнены хромосомными аберрациями плода, которые в подавляющем большинстве случаев ведут к спонтанным абортам.
Диагноз ряда наследственных болезней не представляет существенных затруднений и основывается на данных, полученных в результате общего клинического обследования (например, диагноз болезни Дауна, гемофилии, гаргоилизма, адреногенитального синдрома). Однако в большинстве случаев при диагностике наследственных болезней возникают серьезные затруднения в связи с тем, что многие из этих болезней по клиническим проявлениям очень сходны с приобретенными болезнями— так называемые фенокопиями наследственных болезней. Известно также существование ряда фенотипически сходных, но гетерогенных в генетическом отношении болезней (например, синдром Марфана и гомоцистинурия, галактоземия и синдром Лоу, фосфат-диабет и почечный канальцевый ацидоз). Все случаи атипично протекающих или хронических заболеваний требуют клинико-генетического анализа. На наследственную болезнь может указывать наличие специфических клинических признаков. Среди них особое диагностическое значение могут иметь признаки дисплазии— эпикант, гипертелоризм, седловидный нос, особенности строения лица («птичье», «кукольное», олигомимичное лицо и др.), черепа (долихоцефалия, брахицефалия, плагиоцефалия, «ягодичная» форма черепа и др.), глаз, зубов, конечностей и др.
При подозрении на наследственных болезни генетическое обследование больного начинают с получения подробных клинико-генеалогических данных на основании результатов его опроса о состоянии здоровья ближайших и отдаленных родственников, а также специального обследования членов семьи, что позволяет составить медицинскую родословную больного и определить характер наследования патологии. Вспомогательное, а в ряде случаев решающее диагностическое значение имеют данные, полученные с помощью различных параклинических методов, в т.ч. биохимических, и цитогенетических и молекулярно-генетических исследований. Разработаны биохимические методы диагностики нарушений обмена веществ, основанные на применении хроматографии, электрофореза, ультрацентрифугирования ит.д. Для диагностики заболеваний, вызванных недостаточностью ферментов, применяют методы определения активности этих ферментов в плазме и клетках крови, в материале, полученном при биопсии органов. Биохимические исследования проводят также на клетках, выращенных в культуре, что позволяет обнаружить ряд болезней, обусловленных нарушением обмена липидов, гликозаминогликанов (мукополисахаридов), углеводов, аминокислот, нуклеиновых кислот,— всего более 100заболеваний. Наиболее разработанными являются методы диагностики болезни Тея— Сакса, синдрома Леша— Найхана, некоторых мукополисахаридов. Проведение биохимических исследований при наследственных болезнях обмена в ряде случаев требует применения нагрузочных проб соединениями, метаболизм которых, как предполагают, нарушен.
Однако сложные аналитические методы не могут быть использованы для массовых обследований. В связи с этим при массовых осмотрах проводят двухэтапное обследование с применением на начальном этапе простых полуколичественных методов и (при положительных результатах обследования на первом этапе)— более сложных аналитических методов, выполнение которых возможно лишь в специальных центрах. Эти программы получили название просеивающих, или скрининг-программ. Внедрение методов автоматического биохимического анализа облегчает проведение массового обследования детей на наследственные болезни Массовые обследования детских контингентов (особенно контингентов новорожденных) позволяют выявлять наследственные нарушения обмена в доклинической стадии, когда диетотерапия и соответствующее медикаментозное лечение способны полностью предупредить развитие тяжелой инвалидности.
Разработка новых методов культивирования клеток, биохимические и цитогенетическое исследования сделали возможной пренатальную диагностику многих наследственных болезней, в т.ч. всех хромосомных болезней и болезней, сцепленных с Х-хромосомой, а также целый ряд генетически обусловленных нарушений обмена веществ. Результаты подобных исследований могут служить показанием для прерывания беременности или начала лечения аномалий обмена еще во внутриутробном периоде. Пренатальная диагностика наследственных болезней показана в тех случаях, когда у одного из родителей обнаруживается структурная перестройка хромосом (транслокация, инверсия), когда возраст беременных женщин превышает 35лет и когда в семье прослеживаются доминантно наследуемые заболевания или существует высокий риск возникновения рецессивных наследственных болезней— аутосомных или сцепленных с Х-хромосомой. Для антенатальной диагностики используют также рентгенографию и ультразвуковое исследование плода.
Лечение и профилактика наследственных болезней. В большинстве случаев наследственных болезней проводится симптоматическое лечение, направленное на коррекцию отдельных проявлений. Одним из наиболее распространенных методов патогенетического лечения наследственных болезней обмена является диетотерапия. Проведение диетотерапии требует строгого соблюдения ряда условий: точного диагноза аномалии обмена, исключающего ошибки, связанные с существованием фенотипически сходных синдромов; максимальной адаптации диеты к потребностям растущего организма; тщательного клинического и биохимического контроля.
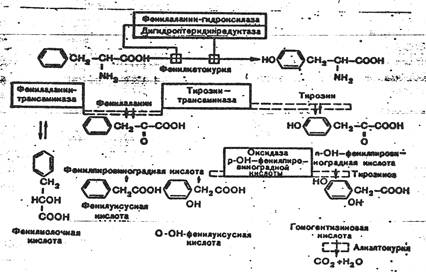
Наиболее полно изучены возможности диетической коррекции обмена фенилаланина при фенилкетонурии. Для диетической коррекции галактоземии созданы специальные препараты. Препараты типа энпита с успехом применяют при лечении других Н.б. (синдрома Марфана, синдрома Лоренса— Муна— Бидля). Предложены также специальные диеты для лечения гистидинемии, гомоцистинурии, кетоацидурии и др.
Продолжается поиск методов лечения больных с наследственными ферментопатиями. Заместительная терапия, при ферментопатиях, в основе которых лежит недостаточность ферментов или их проферментов, участвующих в процессах расщепления и всасывания (муковисцидозе, недостаточности дисахаридаз, трипсиногена и др.), заключается в приеме желудочного сока, препаратов пепсина, трипсина, панкреатина; проходит клинические испытания использование лактазы, дрожжевой сахаразы, гамма-амилазы при нарушениях всасывания лактозы, сахарозы и крахмала,
Заместительная терапия препаратами гамма-глобулина, обогащенными антителами или иммуноглобулинами других классов, проводится при лечении наследственных иммунопатологий, связанных с дефицитом иммуноглобулинов. Для лечения наследственных эндокринных заболеваний вводят кортикостероиды (например, при адреногенитальном синдроме), препараты гормонов щитовидной железы (при гипотиреозе), инсулин (при сахарном диабете) ит.п.
Основным препятствием при лечении наследственных ферментопатий методом введения недостающих ферментов, т.е. методом заместительной ферментотерапии, являются иммунные реакции на введение чужеродного белка. Новые возможности в этом направлении открывает использование искусственно созданных липидных частиц— липосом. Клетки тканей захватывают липосомы, под действием клеточных липаз оболочка липосомы разрушается и фермент получает возможность проявить свое действие внутри клетки. В качестве оболочки для вводимого с терапевтической целью фермента используют также тени эритроцитов больного, капсулы из нейлона. Новым направлением в лечении наследственных болезней является разработка методов индуцирования синтеза ферментов с помощью химических препаратов и гормонов. Так, установлен наследственных болезней но, например, что барбитураты индуцируют синтез глюкуронилтрансферазы— фермента, необходимого для образования глюкуронидов билирубина (так называемого прямого билирубина), стероидных гормонов и ряда других соединений. Отмечено значительное повышение активности глюкуронилтрансферазы под влиянием фенобарбитала у больных с синдромом Криглера— Найяра, который характеризуется резкой гипербилирубинемией в связи с генетически обусловленной недостаточностью этого фермента. Глюкокортиконды активизируют синтез глюкозо-6-фосфат— дегидрогеназы и могут использоваться при лечении гликогеноза I типа (болезни Гирке) с целью предупреждения гипогликемических состояний и снижения интенсивности накопления гликогена в тканях. Установлено индуцирующее влияние кортикостероидов на синтез и созревание ферментных систем кишечника, в частности дисахаридаз. Эстрогенные гормоны обусловливают увеличение концентрации церулоплазмина в крови, поэтому их используют при лечении гепатоцеребральной дистрофии.
Индуцировать синтез ферментов могут и витамины, причем это особенно заметно при так называемой витаминозависимых состояниях, которые характеризуются развитием гипо - или авитаминоза не в связи с ограниченным поступлением витаминов в организм, а в результате нарушения синтеза специфических транспортных белков или апоферментов. Хорошо известна эффективность высоких доз витамина В6 (от 100мг и выше в сутки) при так называемых пиридоксинзависимых состояниях и заболеваниях (цистатионинурии, гомоцистинурин, семейной гипохромной анемии, синдроме Клаппа— Комроуэра, болезни Хартнупа, некоторых формах бронхиальной астмы). Высокие дозы витамина D (до 50000— 200000ME в сутки) оказались эффективными при наследственных рахитоподобных заболеваниях (фосфат-диабете, синдроме де Тони— Дебре— Фанкони, почечном канальцевом ацидозе). ВитаминС в дозах до 1000мг в сутки применяют при лечении алкаптонурии. Высокие дозы витаминаА назначают больным с синдромами Гурлер и Гунтера (мукополисахаридозы). Отмечено улучшение состояния больных мукополисахаридозами под влиянием преднизодона.
Успехи пластической и восстановительной хирургии определили высокую эффективность хирургического лечения наследственных и врожденных пороков развития. В практику лечения наследственных болезней внедряются методы трансплантации, что позволит не только заменить органу, подвергшиеся необратимым изменениям, но и осуществлять пересадки с целью восстановления синтеза белков и ферментов, отсутствующих у больных. Большой интерес может представить трансплантация иммунокомпетентных органов (вилочковой железы, костного мозга) при лечении разных форм наследственных иммунопатологий.
Одним из методов лечения наследственных болезней является назначение препаратов, связывающих токсические продукты, образующиеся в результате блокирования определенных биохимических реакций. Так, для лечения гепатоцеребральной дистрофии (болезни Вильсона— Коновалова) применяют препараты, образующие растворимые комплексные соединения с медью (унитиол, пеницилламин). Комплексоны, специфически связывающие железо, находят применение при лечении гемохроматоза, а комплексоны, образующие растворимые комплексные соединения кальция,— при лечении наследственных тубулопатий с нефролитиазом. При лечении гиперлипопротеинемий применяют холестирамин, который связывает холестерин в кишечнике и препятствует его реабсорбции.
Похожие работы
... , но и в обычных условиях жизни у практически здоровых людей , уделяющих недостаточное внимание разнообразию пищевого рациона . Развитию этой формы витаминной недостаточности способствуют широкое использование в питании рафинированных продуктов , лишенных витаминов в процессе их производства ( хлеба тонкого помола , сахара и др.); потеря витаминов при длительном хранении и неправильной кулинарной ...
... разработке? ) генетической инженерии для лечения наследственных болезней, даже если будут сделаны решительные прорывы в синтезе соответствующих генов и способах их доставки в клетки-мишени. Генетика человека еще не располагает достаточными сведениями обо всех особенностях функционирования генетического аппарата человека. Пока еще неизвестно, как он будет работать после введения дополнительной ...
... , в известной степени, определяется генотипом. Кроме хромосомной, различают внехромосомную, или цитоплазматическую, наследственность. Хромосомная наследственность связана с распределением носителей наследственности (генов) в хромосомах. Цитоплазматическая наследственность проявляется в наследовании признаков, которые контролируются внехромосомными, цитоплазматическими наследственными факторами, ...
... Сфингомиелиназа Сфингомиелин, холестерин. Лейкоциты, материал биопсии органов, фибробласты кожи (инфантильная и ювенильная формы) Материал биопсии Гиперлипопротеидемии (ГЛП) Наследственные заболевания нарушений липидного обмена представлены большим количеством нозологических форм, которые обусловливают развитие атеросклероза и других форм сердечно-сосудистой патологии. Впервые ...
0 комментариев