Свободные аминокислоты нервной системы
Содержание
Введение
1. Содержание, локализация и транспорт аминокислот
2. Метаболизм дикарбоновых аминокислот и глутамина
3. Глутамат и аспартат
4. N-Ацетиласпарагиновая кислота
5. Гамма-аминомасляная кислота
6. Компартментализация метаболизма аминокислот
7. Глицин и пути его обмена
8. Серусодержащие аминокислоты
9. Ароматические аминокислоты нервной ткани и их метаболизм
10. Основные аминокислоты
Выводы
Введение
Свободные аминокислоты нервной ткани или так называемый аминокислотный пул на протяжении многих лет были объектом тщательного изучения. Это объясняется не только исключительной ролью аминокислот как источников синтеза большого числа биологически важных соединений, таких, как белки, пептиды, некоторые липиды, ряд гормонов, витаминов, биологически активных аминов и др. Аминокислоты или их дериваты участвуют и в синаптической передаче, в осуществлении межнейрональных сетей в качестве нейротрансмиттеров и нейромодуляторое. Существенной является также их энергетическая значимость, ибо аминокислоты глутаминовой группы непосредственно связаны с циклом трикарбоновых кислот.
1. Содержание, локализация и транспорт аминокислот
Транспорт аминокислот в мозг и из мозга, скорости их метаболических превращений, включения в белки и катаболизма определяют их концентрацию в этом органе. Состав пула свободных аминокислот при нормальных физиологических условиях довольно стабилен и характерен для мозга. Он лишь незначительно варьирует в мозге различных видов позвоночных. Нервная ткань обладает уникальной способностью поддерживать относительное постоянство уровней аминокислот при различных физиологических и даже некоторых патологических состояниях. Аминокислотный фонд мозга человека составляет в среднем 34 мкмоль на 1 г ткани, что значительно превышает их содержание как в плазме крови, так и в спинномозговой жидкости.
Характерны высокая концентрация глутаминовой кислоты, глутамина, аспарапшовой, N-ацетиласпарагиновой и у-аминомасляной кислот, а также их интенсивный метаболизм. Эти пять аминокислот составляют 75% фонда всех свободных аминокислот головного мозга, причем ГАМК и N-ацетиласпарагиновая кислоты локализованы почти исключительно в нервной ткани. Высокие концентрации дикарбоновых аминокислот и глутамина обнаружены в мозге всех изученных видов животных.
Таблица 1
Содержание свободных аминокислот в мозге, плазме крови и спинномозговой жидкости человека
| Аминокислоты | Мозг | Плазма крови | СМЖ | |||
| Глугаминовая | 10,6 | 75% | 0,05 | 23% | 0,225 | 60% |
| N-Ацетиласпарагиновая | 5,7 | - | ||||
| Глутамин | 4,3 | 0,70 | 0,030 | |||
| ГАМ К | 2,3 | - | - | |||
| Аспарагиновая | 2,2 | 0,01 | 0,007 | |||
| Цистатионин | 1,9 | 25% | - | 77% | - | 40% |
| Таурин | 1,9 | 0,10 | - | |||
| Глицин | 1,3 | 0,40 | 0,013 | |||
| Алании | 0,9 | 0,40 | 0,017 | |||
| Глутатион | 0J | ОДО | 0,010 | |||
| Серин | 0,7 | 0,10 | 0,010 | |||
| Треонин | 0,2 | 0,15 | 0,025 | |||
| Триптофан | 0,05 | 0,05 | 0,010 | |||
| В алии | 0,2 | 0,25 | 0,013 | |||
| Лизин | 0,1 | 0,12 | 0,014 | |||
| Лейцин | 0,1 | 0,15 | 0,004 | |||
| Пролин | 0,1 | ОДО | - | |||
| Аспарагин | 0,1 | 0,07 | - | |||
| Метионин | од | 0,02 | 0,003 | |||
| Изолейцин | 0,1 | ОДО | 0,080 | |||
| Аргинин | 0,1 | ОДО | 0,060 | |||
| Цистеин | 0,1 | ОДО | 0,002 | |||
| Фенил аланин | ол | ОДО | 0,010 | |||
| Тирозин | 0,1 | 0,10 | 0,006 | |||
| Гистидин | од | ОДО | 0,003 | |||
Постоянство суммарного аминокислотного пула головного мозга сопровождается региональной неоднородностью их содержания, что отражает морфологическую, физиологическую и функциональную гетерогенность этого органа. Наиболее неравномерно распределены аминокислоты, выполняющие функцию нейротрансмиттеров, такие, как глутаминовая кислота, таурин, ГАМК, глицин и др.
Таблица 2
Содержание аминокислот в различных областях мозга кошки
| Аминокислоты | Тал a wye | Средний мозг | Мозолистое тело | Кора височной доли | Мозжечок |
| Глутаминовая | 12,36 | 9,71 | 10,58 | 12,93 | 12,63 |
| Аспарагиновая | 2,71 | 4,06 | 1,41 | 3,09 | 2,85 |
| Таурин | 1,06 | 1,62 | 2,99 | 1,89 | 3,12 |
| Глицин | 1,72 | 2,77 | 0,614 | 1,25 | 1,49 |
| Алании | 0,591 | 1,09 | 0,704 | 0,863 | 0,895 |
| ГАМК | 3,65 | 5,81 | 0,961 | 1,39 | 1,49 |
| Тирозин | 0,05 | 0,059 | 0,049 | 0,039 | 0,06 |
| Валин | 0,145 | 0,152 | 0,096 | 0,117 | 0,097 |
| Лизин | 0,278 | 0,379 | 0,268 | 0, 194 | 0,219 |
Различные органеллы клеток головного мозга контролируют уровень аминокислот, накапливая их часто против концентрационных градиентов.
Постоянство качественного и количественного состава аминокислот в метаболических фондах мозга обеспечивается такими взаимосвязанными процессами, как поступление аминокислот из циркулирующей крови, отток их из мозга в кровь и участие в реакциях внутриклеточного метаболизма. В организме все эти процессы сбалансированы слаженным функционированием гомеостатических механизмов, гематоэнцефалического барьера и мембранным транспортом.
Транспорт аминокислот в мозг - многоступенчатый процесс. Прежде всего происходит транспорт через гематоэнцефалический барьер, локализованный в эндотелии мозговых капилляров, затем осуществляется транспорт из внеклеточной жидкости в клетки мозга, а далее - в субклеточные органеллы. Существуют системы активного транспорта аминокислот не только в мозг, но и из него, - обе они энергозависимы.
Исследование конкурентных отношений в транспорте аминокислот выявило наличие восьми классов транспортных систем, которые существуют для аминокислот с родственной структурой и зависят от ионного заряда и размеров их молекул. В ряде случаев одна аминокислота может транспортироваться с участием нескольких транспортных систем, выбор той или иной системы определяется составом аминокислотного пула. Для мембранного транспорта аминокислот характерен ряд особенностей: а) перенос аминокислот часто происходит против высоких концентрационных градиентов; б) этот процесс энергозависим; в) на него влияют температура и рН среды; г) он ингибируется анаэробиозом и ферментными ядами; д) перенос аминокислот связан с активным мембранным транспортом ионов, в частности, он Na-зависим; е) обнаружено конкурентное торможение мембранного транспорта одних аминокислот другими и др. Такие конкурентные взаимодействия играют важную роль в патологии, когда изменяется уровень индивидуальных аминокислот в крови. Ниже мы приведем примеры таких патологических состояний.
Уровень специфичности транспортных систем для разных аминокислот неодинаков. Особенно велика специфичность и мощность систем для аминокислот, выполняющих роль нейротрансмиттеров. Эти системы не только обеспечивают пластические и энергетические нужды клетки, но служат такие для специфического процесса быстрого снижения концентрации нейротрансмиттера в зоне синоптической щели. Высокоизбирательное поглощение нейротрансмиттера осуществляется как пресинаптической областью, так и клетками окружающей глии.
Еще один своеобразный механизм транспорта аминокислот связан с метаболизмом широко распространенного во всех тканях, в том числе и в нервной, трипептида глутатиона, цикл синтеза и деградации которого известен под названием у-глутамильного цикла. Наиболее интересным и ключевым ферментом этого цикла является у-глуталшлтранспептидаза, прочно связанная с клеточной мембраной. Этот энзим способен переносить у-глутамильную группу глутатиона, находящегося внутри клетки, на аминокислоту, локализованную с наружной стороны мембраны, и переносить образующийся дипептид внутрь клетки. Следующий фермент этого цикла - у-глутамилциклотрансфераза высвобождает аминокислоту. Таким образом, у-глутамил транспептидазная реакция является одним из механизмов транспорта аминокислот внутрь клетки.
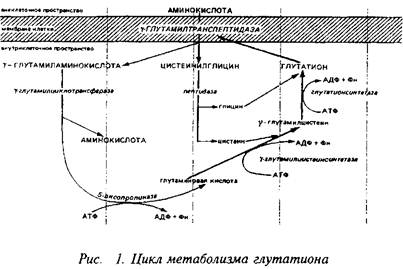
При нормальных условиях скорость транспорта аминокислот не лимитирует непосредственно их метаболизм, так как скорости синтеза и деградации ниже скорости транспорта. Поэтому аминокислоты и аккумулируются мозгом, формируя пул свободных аминокислот. Без пополнения извне пул свободных аминокислот довольно быстро истощается. Так, количество аминокислот, которое используется для синтеза белков мозга, нейропептидов и нейромедиаторов в течение 30 мин, равно общему церебральному пулу большинства свободных аминокислот.
Активность систем транспорта аминокислот, так же как и состав их пула, изменяется в процессе развития мозга. Аминокислоты проникают в мозг молодых животных быстрее и достигают более высоких концентраций, чем у взрослых.
В литературе отсутствуют сообщения о болезнях, вызванных нарушением транспорта аминокислот в мозг, вероятно, потому, что они летальны. Даже дефекты транспорта аминокислот в другие ткани ведут к заболеваниям, имеющим неврологические последствия.
Наряду с неопасным для жизни синдромом Хартнупа, вызванным дефектом транспорта триптофана в малый кишечник и почки и схожим клинически с пеллагрой, известен ряд недугов с тяжелыми неврологическими последствиями, также обусловленных дефицитом поступления аминокислот. Среди них - цистиноз - нарушение транспорта цистина в клетки, особенно почек; цисти-ноз сопровождается фотофобией и повреждением глаз. Тяжелым, нередко летальным заболеванием, связанным с транспортом аминокислот в кишечник, является окулоцеребральный синдром. Он сопровождается глаукомой, катарактой, слепотой. Перечень этих болезней, вызванных нарушением транспорта триптофана, метионина, нейтральных и других аминокислот в кишечнике и других органах, довольно велик, причем все они косвенно затрагивают уровень аминокислот в мозге и имеют поэтому неврологические проявления.
2. Метаболизм дикарбоновых аминокислот и глутамина
Более 2/3 аминоазота аминокислот приходится на долю глутамата и его производных; эти аминокислоты доминируют в количественном отношении в мозге всех изученных видов животных. В спинном мозге наблюдается аналогичная картина, а периферическая нервная система содержит значительно меньше глутамата, глутамина, N-ацетиласпартата, чем головной мозг, а ГАМК почти отсутствует в периферических нервах позвоночных. При высоком уровне этих аминокислот в головном мозге метаболизм их также чрезвычайно быстрый.
3. Глутамат и аспартатОсобенностью метаболизма глутамата в нервной ткани является его тесная связь с интенсивно функционирующим в этом органе циклом трикарбоновых кислот, что и позволяет считать его промежуточным продуктом энергетического метаболизма. Так, уже через 30 мин после инъекции меченой глюкозы более 70% радиоактивности растворимой фракции приходится на долю глутамата и его производных. Этому способствует чрезвычайно быстрое взаимопревращение глутамата и а-кетоглутарата в ЦНС. Высокий процент включения радиоактивности из глюкозы в аминокислоты мозга явился основанием для предположения, что утилизация глюкозы в этом органе в значительной степени происходит через биосинтез и окисление аминокислот.
Непосредственным предшественником для синтеза глутамата в мозге является а-кетоглутаровая кислота, которая может превращаться в глутамат или путем прямого восстановительного аминирования с участием глутаматдегидрогеназы, или путем переаминирования.
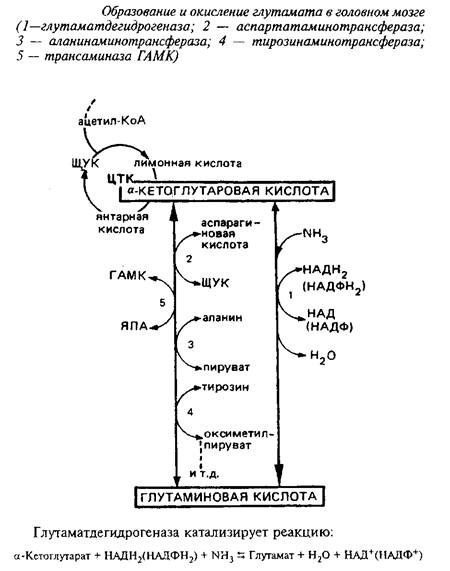
Энзим менее активен в мозге, чем в печени, присутствует в митохондриях, требует в качестве кофакторов пиридиннуклеотидов и активируется АДФ. Км этого энзима для аммония близок к 8 мМ. Реакция обратима, однако равновесие сильно сдвинуто в сторону прямой реакции, т.е. синтеза глутаминовой кислоты.
Таким образом, в головном мозге глутаматдегидрогеназная реакция участвует не столько в окислении глутамата, сколько в синтезе его из а-кетоглутаровой кислоты, обеспечивая тем самым непрерывное превращение свободного аммиака в аминоазот аминокислот. Основной же путь окисления глутамата в мозге - через переаминирование.
В митохондриях мозга 90% глутамата подвергается переаминированию с образованием аспартата. Фермент, катализирующий переаминирование глутамата с щавелевоуксусной кислотой, - аспартатаминотрансфераза является наиболее мощной трансаминазой головного мозга. Выделены два изоэнзима аспартатаминотрансферазы, локализованных в митохондриях и цитоплазме. Функциональная роль их различна. Митохондриальный фермент связан в основном с функционированием ЦТК, цитоплазматический определяет интенсивность глюконеогенеза.
Как уже отмечалось, путь метаболизма глутамата через переаминирование намного активнее дегидрогеназного. В регуляции соотношения между этими двумя путями, конкурирующими за один субстрат, важная роль принадлежит макроэргическим соединениям. В интактных митохондриях энзим взаимодействует по преимуществу с НАДФ+ и интенсивность реакции пропорциональна отношению НАДФ+/НАЦФН2. Макроэргические соединения способствуют превращению НДЦФ+ в НАДФН2 и тем самым подавляют дезаминирование глутамата. Наоборот, трансаминазный путь требует расходования макроэргических соединений. Поэтому выбор между этими двумя реакциями определяется энергетическими возможностями митохондрий.
При нормальном функционировании ЦТК дегидрогеназный путь окисления глутамата подавлен, а трансаминазный активно протекает. В результате уменьшения количества макроэргических соединений, например при добавлении к митохондриям разобщителя окислительного фосфорилирования 2,4-динитро-фенола, подавляется трансаминазный путь при одновременном резком усилении дегидрогеназного пути окисления глутамата.
Взаимопревращение а-кетоглутарата и глутамата происходит чрезвычайно быстро. В мозге был идентифицирован метаболический путь такого взаимопревращения, получивший название аспартат-малатного шунта, служащего для транспорта восстановительных эквивалентов из цитозоля в митохондрии.
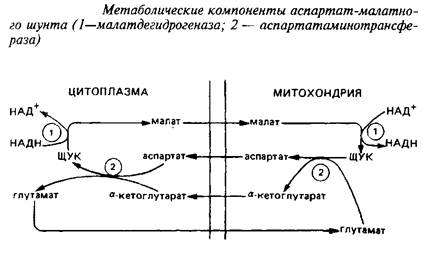
Уже упоминалось, что различные органеллы клеток мозга могут индивидуально контролировать уровни аминокислот, накапливая их против градиента концентрации. Примером этого могут служить изолированные из ЦНС митохондрии, которые быстро поглощают глутамат и малат, освобождая соответствующие количества аспартата и а-кетоглутарата. Это означает, что ток аспартата через митохондриальную мембрану связан с током глутамата в обратном направлении; также реципрокно связаны ток малата и а-кетоглутарата. Энзимы, катализирующие отдельные реакции малат-аспартатного шунта, превалируют в тканях ЦНС. В нейронах малат-аспартатный шунт является преобладающим механизмом переноса восстановительных эквивалентов в митохондрии.
Таким образом, глутаминовая кислота выполняет чрезвычайно важную функцию в энергетическом обеспечении головного мозга, которая заключается в поддержании метаболитов ЦТК на определенном и довольно высоком уровне, а также в снабжении митохондриальных синтетических процессов восстановительными эквивалентами.
Большое значение имеет образование аммиака из глутамата. В головном мозге обнаружены многочисленные аминотрансферазы основных, кислых, нейтральных и ароматических аминокислот. При участии этих ферментов аминогруппы различных аминокислот переносятся в конечном счете на глутамино-вую кислоту. Последняя переаминируется с ЩУК при участии аспартатами-нотрансферазы с образованием аспартата. Образование аммиака из аспартата происходит различным образом в митохондриях и цитоплазме. В митохондриях этот процесс связан с аминированием дезаминоформ НАД+ и включает в себя три ферментативных реакции.
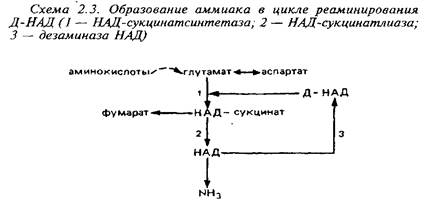
Вне митохондрий действует другой циклический процесс образования аммиака, в котором аспартат реаминирует инозинмонофосфат.
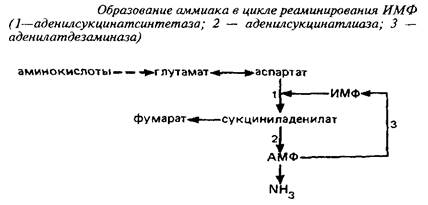
Для удаления аммиака в ЦНС служит глутаминсинтетазная реакция.
Глутаминсинтетаза катализирует реакцию:
![]()
Этот энзим в мозге животных находится в более высокой концентрации, чем в других органах, составляя 0,2% от общего белка мозга. Энзим требует АТФ и Mg+ и подавляется глицином и аланином. Км для аммония - порядка 0,39 мМ, т.е. при нормальной концентрации аммония в мозге фермент работает в режиме полунасыщения. В нормальных физиологических условиях, когда имеется достаточный уровень АТФ, глу-таминсинтетазная реакция направлена в сторону связывания аммиака.
Образование глутамина является важным механизмом детоксикации аммония, к которому мозг чрезвычайно чувствителен и накопление которого губительно для ЦНС. В частности, повышение аммиака в мозге до концентрации 0,6 мМ сопровождается судорогами. Системное введение солей аммония вызывает конвульсии и увеличение содержания глутамина в мозге. В случае серьезных повреждений печени повышается концентрация аммония и глутамина в спинномозговой жидкости - в этих случаях наблюдается кома. Симптомы печеночной комы смягчаются введением глутамата. Основная часть глутаминсинтетазы локализована в глиальных клетках и лишь небольшая часть ее представлена в нервных окончаниях.
Дезаминирование глутамина катализируется глутаминазой, ферментом, наиболее активным в нейронах, где он локализован в митохондриях. Следует отметить, что активность этого фермента в головном мозге невелика; продукты реакции - глутаминовая кислота и аммоний - тормозят активность фермента.
Предполагается участие этого фермента в мембранном транспорте глутамата. Известно, что биологические мембраны более проницаемы для глутамина, чем для глутамата, и глутаминаза может участвовать в превращении глутамина крови во внутриклеточный глутамат. Глутаминаза играет важную роль также в регуляции содержания глутамата в нервных окончаниях. Тот факт, что глутаминсинтетаза локализована в основном в глиальных клетках, а глутаминаза наиболее активна в нейронах, а также то, что глутамин оказался главным предшественником глутамата и ГАМ К, выполняющих трансмиттерную функцию, послужил основанием для концепции о существовании глушаминового цикла, Глутамат, поглощаясь глиальными клетками, превращается в глутамин в синтетазной реакции, последний входит в нейроны, образуя там глутаминовую кислоту. Таким образом, глутамин служит глиально-нейронааьным транспортером глутамата.
Другой важной функцией глутамата является его участие в синтезе белков и биологически активных пептидов. Глутамат и глутамин составляют вместе от 8 до 10% общих аминокислотных остатков в гидролизате белков мозга. В частности, два хорошо изученных мозгоспецифичных белка - S-100 и 14-3-2 - содержат особенно высокую долю глутаминовой кислоты. Глутамат является также составной частью ряда малых и средних регуляторных пептидов мозга. Это прежде всего глутатион и ряд у-глутамильньгх дипептидов. Некоторые нейропептиды содержат циклическое производное глутамата - аироглутамат в качестве N-терминального остатка, который предохраняет эти пептиды от протеолиза. К таким "пептидам относятся люлибе-рин, тиролиберин, нейротензин, бомбезин и др. .
Введение глутамата в различные районы мозга приводит либо к судорожной активности, либо к распространяющейся депрессии, даже если количество его мало по сравнению с нормальной концентрацией глутамата в мозге. Глутамин не вызывает такого эффекта. При внутривенном введении глутамат может вызвать гибель клеток в определенных районах ЦНС, особенно вокруг желудочков мозга, где менее развит гематоэнцефалический барьер. Нейроны незрелых животных, у которых еще отсутствует высокоразвитый гематоэнцефалический барьер, также очень чувствительны к глутамату. Оральное введение больших количеств глутамата не действует на ЦНС большинства людей, а соли глутамата широко используются в качестве пищевой приправы. Однако у некоторых лиц обнаруживается повышенная чувствительность к глутамату натрия, он вызывает сенсорные и моторные нарушения, включая ощущение жжения, напряжение лица, боль в грудной клетке и головную боль. Эти симптомы известны как "синдром китайских ресторанов", так как глутамат натрия широко используется в китайской кухне. Многие аналоги глутамата токсичны.
Остановимся на некоторых сторонах нейротрансмиттерной функции глутамата. Для того чтобы глутамат эффективно функционировал в качестве нейротрансмиттера, его модальная внеклеточная концентрация должна быть ниже той, которая вызывает деполяризацию мембран. В действительности она колеблется от 1 до 10 мкМ; такая низкая внеклеточная концентрация глутамата поддерживается активным транспортом в нейроны и особенно в глиальные клетки.
В процессе выхода глутамата в синаптическую щель концентрация его там значительно повышается - до 1 мМ.
Последующий обратный захват глутамата нейронами и астроцитами осуществляется с участием Na-зависимых высокоаффинных переносчиков, из синаптической щели глутамат удаляется в основном путем захвата астроцитами. Для функционирования глутамата в качестве нейротрансмиттера необходимо постоянное пополнение его пула в нервных окончаниях.
Предшественниками трансмиттерного пула глутамата могут быть глюкоза и а-кетоглутарат.
Глутамат может также образовываться из орнитина и аргинина. Но основным источником нейротрансмиттерного глутаматного пула, по данным изотопных исследований, оказался глутамин, который синтезируется в основном в астроцитах, где локализована глутаминсинтетаза.
Далее он легко транспортируется через мембрану астроцитов и с помощью активных переносчиков достигает нервных окончаний.
4. N-Ацетиласпарагиновая кислотаОдним из доминирующих компонентов пула свободных аминокислот мозга является N-ацетиласпарагиновая кислота
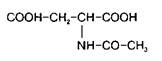
Ее концентрация у большинства видов животных в два раза превышает таковую аспарагиновой кислоты. В ненейрональной ткани обнаружены только следы АцА. Она находится в более высокой концентрации в сером веществе по сравнению с белим, представлена также в периферической нервной системе, в сетчатке. Ее концентрация низка при рождении и повышается в процессе развития животного.
АцА образуется с участием ацетил-КоА. Энзим, катализирующий эту реакцию, очищен и изучен. Точная функция АцА в мозге еще не ясна, хотя имеются предположения, что она является частью внутриклеточного фиксированного пула анионов или резервуаром ацетильных групп, а также источником N-ацетилированных конечных групп для синтеза определенных бел-
ков и пептидов мозга. Показано, что ацетильные группы экзогенной АцА кислоты служат предпочтительным источником углерода для синтеза жирных кислот в развивающемся мозге. В головном мозге оказалось два пространственно разобщенных фонда АцА: малый, высокоактивный, локализованный в глии, и большой, медленно обменивающийся, - в нейронах.
5. Гамма-аминомасляная кислота
Одним из главных компонентов пула свободных аминокислот головного мозга различных животных является у-аминомасляная кислота, продукт а-декарбоксилирования глута-миновой кислоты. Цикл превращений ГАМК в мозге включает три сопряженных энзиматические реакции, получившие название ГАМК-шунта.
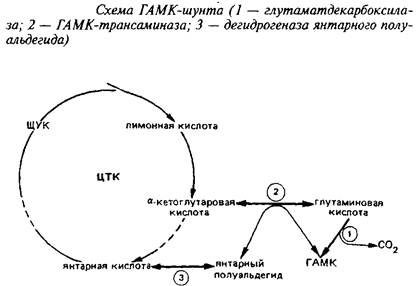
Он является ответвлением ЦТК на участке от а-кетоглутарата до сукцината. При участии фермента i лугам атдекарбокс ил азы отщепляется первый карбоксил L-глутаминовой кислоты с образованием ГАМК.
Этот энзим присутствует только в ЦНС и главным образом в сером веществе. ГДК синтезируется в нейрональной соме, а затем очень быстро транспортируется вдоль аксона. ГДК нуждается в пиридоксальфосфате в качестве кофактора, как большинство других декарбоксилаз аминокислот. Кофактор прочно связан с энзимом. Молекулярная масса энзима 85 кД, Kw для глутамата около 0,7 мМ, а Км для пиридоксальфосфата 0,05 М. ГДК специфичен для глутамата, слабо взаимодействует с аспарагиновой кислотой. Скорость ГДК-реакции - лимитирующая ступень синтеза ГАМК. Уровень ГАМК в различных областях нервной системы регулируется действием ГДК и при нормальных условиях мало зависит от действия энзимов деградации ГАМК. ГДК является маркером ГАМК-ергических синапсов.
Энзимы катаболизма ГАМК локализованы отдельно от ГДК. ГАМК-трансаминаза находится в сером веществе мозга, но встречается также и в других тканях. Она также требует пиридоксальфосфат в качестве кофактора и связана с ним прочно. ГАМК-Т обнаружена в митохондриях, в то время как ГДК и ГАМК локализованы в синаптосомах. Км ГАМК-Т для всех субстратов очень высока.
Конечный энзим шунта - дегидрогеназа янтарного полуальдегида - превращает янтарный полуальдегид в янтарную кислоту. Он распространен в ЦНС там же, где и ГАМК-Т. Это митохондриальный энзим, который специфичен для янтарного полуальдегида и НАД"", активируется сульфгидрнлъными реагентами и подавляется субстратом при концентрации последнего выше 10~М.
ГАМК является наиболее широко распространенным медиатором торможения в нервной системе. У млекопитающих она локализована в нервных окончаниях тормозных нейронов ЦНС. ГАМК тормозит биоэлектрическую активность не только головного мозга позвоночных, но и нервных цепочек и ганглиев беспозвоночных животных. Соответственно ГАМК и ферменты ее обмена также локализованы в нервных структурах беспозвоночных, совпадающих с расположением тормозных синапсов. Физиологическое действие ГАМК обусловлено взаимодействием со специальными рецепторами.
6. Компартментализация метаболизма аминокислотКомпартментализация метаболизма является ключевым фактором взаимоотношений между глутаматом, глутамином и ГАМК, Впервые это явление было открыто в лаборатории Вэлша в конце 50-х - начале 60-х годов и известно под названием эффекта Вэлша. При определенных условиях в опытах с использованием меченых предшественников специфическая радиоактивность продукта, образованного в короткий промежуток времени, превышает специфическую активность предшественника иногда в несколько раз. Эти наблюдения позволяют сделать заключение, что метаболизм имеет место в малом, высокоактивном пуле, а меченый предшественник разбавляется при выделении большим количеством немеченого предшественника из другого, малоактивного пула.
Инъекция меченого глутамата, аммония, бикарбоната, ацетата, бутирата, цитрата и других, как правило, приводила к тому, что специфическая радиоактивность глутамина была выше предшественника, изолированного вскоре после инъекции. Этот эффект не был обнаружен после инъекции меченой глюкозы, пирувата, лактата, глицерина. Данные позволили заключить, что глюкогенные субстраты метаболируют до аминокислот в компартментах, отличных от тех, в которых обмениваются кетогенные субстраты.
Эффект Вэлша - специфическое свойство нервной системы и демонстрируется в опытах как in vitro, так и in vivo. В дальнейшем кинетическими исследованиями с различными метаболическими предшественниками было показано наличие в головном мозге различных метаболических компартментов цикла трикарбоновых кислот и аминокислот, связанных с этим циклом. Некоторые исследователи ограничивают число компартментов двумя - большим и малым, другие описывают до шести метаболических компартментов. Очевидным является факт, что каждый компартмент является суммой большого числа микрокомпартментов с более или менее сходными метаболическими свойствами.
"Большой" компартмент включает в себя относительно большие пулы промежуточных соединений, которые быстро обменивается с большим пулом глутамата и малым пулом глутамина. Глюкоза используется во всех компартментах, но в большой компартмент включается до 90% гликолитического потока и большая часть общего потока через ЦТК. Глюкогенные предшественники метаболируют преимущественно в этом ком-партменте, и глюкоза может рассматриваться как предпочтительный метаболит большого компартмента. Этот же компартмент содержит основную часть общего глутамата и аспартата. Однако скорость синтеза глутамина в нем относительно низка.
Полагают, что "большой" компартмент расположен главным образом в нейронах и участвует преимущественно в энергетических процессах. Предположение о нейрональной локализации "большого" метаболического компартмента подтверждается, например, исследованиями на животных с различным ти-реоидным статусом. Так, удаление щитовидной железы при рождении животного выливается в недоразвитие нейрональных систем. Одновременно наблюдается недоразвитие "большого" метаболического компартмента. Напротив, обработка тиреоид-ными гормонами ускоряет созревание мозга и развитие метаболической компартментации.
"Малый" метаболический компартмент включает в себя ЦТК, но с малыми пулами его компонентов, которые быстро обмениваются с малым пулом глутамата, находящимся, в свою очередь, в равновесии с большим пулом глутамина. "Малый" компартмент является главным источником глутамина. Окислительная способность "малого" компартмента низка; вероятно, он не богат структурами, вовлекаемыми в синтез белка, и митохондриями. Морфологическая характеристика астроглии соответствует биохимическим свойствам "малого" метаболического компартмента. Пул глутамата, связанный с синтезом глутамина, составляющий малую долю общего пула глутамата, находится в астропи-тах. Последние составляют лишь около четверти от общего объема ткани мозга, причем концентрация глутамата в них ниже, чем в ткани ЦНС в целом. Коммуникация между "малым" и "большими" компартментами осуществляется через транспорт глутамина и ГАМК, а также путем аксонального тока белков из нейронального перикариона к нервным окончаниям.
Значение метаболических компартментов состоит в пространственном отделении биосинтетических процессов от тех метаболических путей, которые строго контролируются энергетическими нуждами. Это явление характерно именно для нервной ткани, которая отличается большой функциональной гетерогенностью составляющих ее элементов, большой долей крупных и средних клеток с разнообразными системами органелл и, наконец, большой протяженностью отростков нервных клеток, что затрудняет возможность смешивания метаболитов.
Метаболическая компартментализация аминокислот, в частности аминокислот глутаминовой группы, особенно ярко проявляется в субклеточной локализации ферментов в ГАМК-ергическом нейроне и в астроцитах. Так, пируваткар-боксилаза локализована преимущественно в астроцитах, в то время как пируватдегидрогеназный комплекс более активен в нейронах, чем в астроцитах. Преимущественная локализация пируватдегидрогеназы в нейронах ответственна в конечном счете за низкое включение углерода глюкозы, лактата и глицерина в глутамин. Глутамин-синтетаза преимущественно локализована в астроцитах, а глутаминаза - в нейронах. Глутамат, поглощаясь астроцитами, превращается в глутамин в глутамин-синтетазной реакции. Глутамин, выйдя из астроцитов и входя в нейроны, образует глутамат и далее ГАМК. Таким образом, глутамин и ГАМК осуществляют коммуникацию между большим и малым метаболическими компартментами.
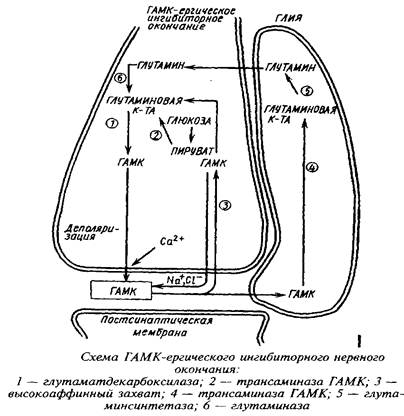
Помимо нейронально-глиального транспорта аминокислот в последние годы установлена возможность перемещения свободных аминокислот от проксимального к дистальному концу нейрона. Так, глутамат, введенный в мозг, передвигается вдоль аксонов двигательных нейронов и оказывается в мышечных нервных окончаниях.
7. Глицин и пути его обменаГлицин участвует не только в биосинтезе белков, но и в других многочисленных биосинтетических процессах, таких, как образование пуринов, порфиринов, креатина, этаноламина, холина, глутатиона и др. Глицин функционирует также в качестве ингибиторного трансмиттера главным образом в спинном мозге.
Так как потребление глицина в нервной ткани относительно велико, а поступление его из крови происходит медленно, значительная часть глицина синтезируется в мозге de novo. Глюкоза и серии являются главными источниками глицина в ЦНС. Серии может образовываться из глюкозы через 3-фосфоглице-риновую кислоту. Кроме того, серии сравнительно быстро поступает из циркулирующей крови. Синтез глицина de novo происходит в нервной ткани из серина путем обратимой N, W-метилентетрагидрофолат-тет-рагидрофолатзависимой трансформации при участии фермента серингидроксил1етилтрансферазы. Реакция катализируется серингидроксиметилтрансферазой и протекает следующим образом:
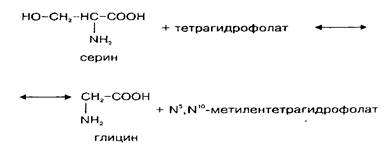
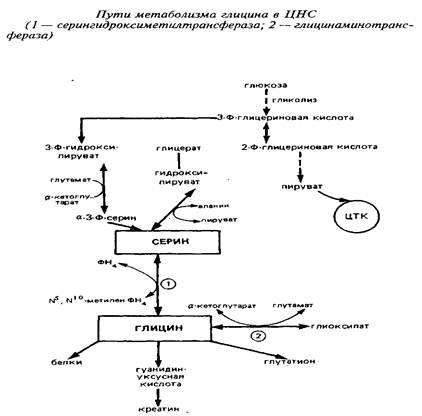
Этот фермент относится к пиридоксальзависимым при оптимальной активности в нем содержится 6 молекул пиридоксальфосфата. Активность фермента в метаболических пулах головного мозга относительно постоянна, высокая активность его обнаружена в спинном мозге и в мозжечке. Активность в сером веществе спинного мозга больше, чем в белом, причем в вентральном сером веществе она значительно выше, чем в дорзальном. Это коррелирует с содержанием глицина.
Другим источником синтеза глицина в нервной системе является глиоксиловая кислота, однако вклад ее в синтез глицина в головном мозге in vivo не может быть значительным, так как ее уровень в мозге низок.
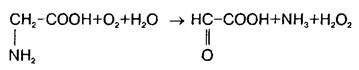
В нервной ткани существует по крайней мере три пути катаболизма глицина. Первый состоит в том, что реакция превращения серина в глицин легко обратима в ткани мозга и серин-гидроксиметилтрансфераза может выступать в качестве энзима деградации глицина. Кроме того, в ЦНС представлены оксидазы аминокислот, которые могут использовать в качестве субстрата наряду с другими аминокислотами глицин:
Третья система распада глицина локализована исключительно в митохондриях и является нетипичной декарбоксилазой аминокислот, так как зависит и от НАД+, и от тетрагидрофолата. Расщепление глицина на одноуглеродные фрагменты протекает по схеме:
![]()
Важно отметить образование в этих реакциях метилентетра-гидрофолата, который может быть использован в мозге как источник одноуглеродных фрагментов. То же следует подчеркнуть применительно к описанной серингидроксиметилтрансферазной реакции.
При участии глицин-расщепляющей системы глицин распадается на метилентетрагидрофолат, диоксид углерода и аммиак, затем происходит окисление метилен-ТГФ с образованием СОз - окончательного продукта распада глицина.
Как уже упоминалось, глицин является ингибиторным трансмиттером в спинном мозге. В других районах депрессорное действие глицина проявляется слабо. Поэтому спинной мозг имеет высокоаффинную и низкоаффинную систему захвата глицина, в то время как кора головного мозга содержит только низкоаффинную систему.
Интересно отметить, что повышенный уровень глинина обнаружен в эпилептогенных районах мозга человека, удаленных хирургическим путем. Он накапливается также в эпилептогенных районах мозга у животных с вызванными кобальтом припадками, причем тяжесть припадков пропорциональна накоплению глицина. Возможно, это - компенсаторные процессы.
Высокий уровень глицина в плазме крови или в моче обычно свидетельствует о нарушении мозговых функций. Гиперглицинемия развивается в раннем возрасте и сопровождается эпизодическими рвотами, подавлением двигательной активности, нарушением ЭЭГ и часто кончается смертью. Известны два типа гиперглииинемии - кетотическая и некетотическая, которая в большинстве случаев тоже летальна. Кетотическая гиперглицинемия сопровождается губчатой дегенерацией белого вещества мозга и задержкой миелинизации.
8. Серосодержащие аминокислотыМетионин представляет особый интерес как источник метильных групп. Полученный с пищей, а также образованный в других тканях, метионин поступает в мозг через систему активного транспорта больших нейтральных аминокислот. Концентрация метионина в целом мозге сравнительно низка - от 10 до 100 нмоль/г сырой массы у различных видов животных. Региональные различия в концентрации метионина невелики. Влияние диеты на концентрацию метионина в мозге также незначительно из-за конкурентных отношений с нейтральными аминокислотами за транспортные системы. Метионин в пуле свободных аминокислот утилизируется на 80% для синтеза белка.
Метаболизм свободного метионина до цистеина начинается с образования S-аденозилметионина, реакция катализируется метионин-аденозилтрансферазой. S-Аденозилметионин является главным донором метальных групп в мозге, необходимых для метилирования катехоламинов, гистамина, фосфатидилэтаноламина, нуклеиновых кислот.
Процессам метилирования отводится важная роль: в проведении сигнала через мембрану, в регулировании жидкостно-сти мембраны и, наконец, в процессах метилирования ДНК. Последние считают вероятными участниками механизмов долговременной памяти. В то время как первая половина цикла превращения метионина связана главным образом с метилированием, вторая часть его ассоциирована в основном с нейротрансмиттерной и нейромо-дуляторной функцией. Оказалось, что 20% серусодержащих аминокислот локализовано в синаптосомах.
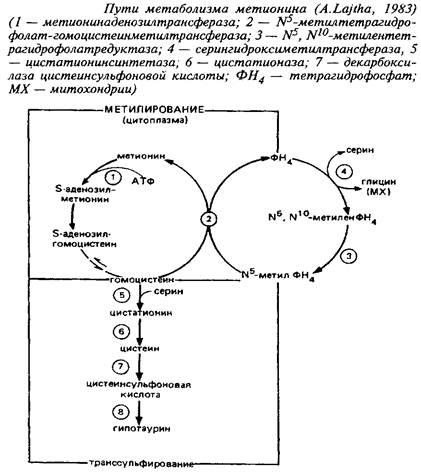
Цистатиония - продукт конденсации гомоцистеина и серина. Фермент, участвующий в этом процессе, - цистатионин-синтаза. Цистатионин является промежуточным продуктом метаболизма таких серусодержащих аминокислот, как метионин, цистеин и таурин. Будучи промежуточным метаболитом в обмене серы, он важен для синтеза сульфатидов и сульфатированных мукополисахаридов. Содержание цистатионина выше в белом веществе, чем в сером.
У человека высокие концентрации цистатионина обнаружены в мозге и гораздо меньшие - в других тканях. Интересно отметить, что мозг человека содержит значительно более высокие концентрации цистатионина, чем мозг животных. Концентрация цистатионина в мозге человека повышается в процессе развития, а в мозге крысы, напротив, снижается. Биологическая роль цистатионина не выяснена. При некоторых психических заболеваниях, а также при действии нейротоксинов содержание цистатионина в мозге резко возрастает. В то же время у некоторых умственно отсталых больных с врожденными нарушениями обмена серусодержащих аминокислот содержание цистатионина в мозге было чрезвычайно низким.
Генетическая потеря цистатионинсинтазы ведет к болезни - гомоцистинурии, которая сопровождается экскрецией гомоцистеина с мочой, повышением содержания гомоцистеина и метионина в крови и дефицитом цистатионина и цистатионинсинтазы в мозге и печени. Гомоцистинурия является второй по распространенности аминоацидурией после фенилкетонурии с ярко выраженным действием на ЦНС. Одной из характеристик болезни является фиброз и утончение кровеносных сосудов. Терапевтическое средство - снижение в диете метионина и доноров метильных групп - таких, как холин. Для таких больных необходимо включение цистина в диету, так как они не могут образовывать его из метионина. Клиническая картина у детей выражается в эпизодических судорожных припадках, тяжелом физическом и умственном отставании.
Таурин образуется в мозге посредством окисления цистеина до цистеинсульфоновой кислоты, которая декарбоксилируется с образованием гипотаурина с последующим окислением его до таурина. Он обнаружен в высоких концентрациях в нервной системе беспозвоночных и позвоночных животных. Высокие концентрации таурина найдены в мозге эмбрионов, а также в ранний период постэмбрионального развития. Так, у мышей в первые дни жизни концентрация таурина выше, чем концентрация аминокислот глутаминовой группы, в 3 раза, а у взрослых это отношение уменьшается.
Региональное распределение таурина неравномерно. Он содержится в нейронах и в глии, причем большая часть его обнаружена в растворимой фракции. В мозге крыс синаптосомальные фракции полосатого тела, коры мозга и мозжечка содержат наиболее высокое количество таурина. Интересно, что таурин - наиболее распространенная аминокислота сетчатки некоторых видов животных.
Подобно другим короткоцепочечным омега-аминокислотам таурин подавляет нейрональную возбудимость, вызывая гиперполяризацию. Таурин - предполагаемый трансмиттер в коре и стволе мозга. По последним сведениям, он может быть нейротрансмиттером в некоторых районах гиппокампа. Инактивация таурина в мозговых синапсах осуществляется с помощью высокоаффинного обратного захвата. Описан также захват таурина глиальными клетками, что указывает на роль глии в модуляции его синаптической функции.
Таурин связан с регуляцией транспорта кальция в нервной ткани. Многие авторы склонны объяснять высокую концентрацию таурина в мозге именно участием его в контроле уровня Са+. Модуляция таурином внутриклеточной концентрации Са+, в свою очередь, регулирует нейрональную возбудимость. Таурин подавляет захват и освобождение Са+ синаптосомами мозга. Более того, он подавляет связывание Са+ микросомами мозга в условиях, стимулирующих деполяризацию. Хотя молекулярный механизм взаимодействия таурина с кальций-регулирующими системами еще не ясен, приведенные данные свидетельствуют о том, что роль таурина в организме не ограничивается только нейромедиаторной функцией.
Интересен факт обнаружения нейропептидов, содержащих таурин, которые оказывают гормоноподобные эффекты.
Таурин является слабым $-адренергическим агонистом, он активирует К+-стимулированное освобождение норадреналина из коры мозга, не влияя на спонтанное освобождение. Интравентрикулярное введение таурина повышает синтез дофамина и норадреналина во всех изученных районах мозга. Влияние его на двигательную активность и регуляцию температуры животного подтверждает медиацию этих эффектов через катехоламинерги-ческую систему. Таурин оказывает антиконвульсивное действие при эпилепсии, блокирует агрессивные реакции у крыс-киллеров. Однако следует иметь в виду, что содержание таурина в мозге трудно корректировать - он плохо проникает через ГЭБ.
Клинически тауриновый дефицит может выражаться в эпилептических припадках, наследственной атаксии Фридрейха, куриной слепоте и др.
9. Ароматические аминокислоты нервной ткани и их метаболизмАроматические аминокислоты - триптофан, фенилаланин и тирозин - важны как предшественники 5-гидрокситрилтамина и катехоламинов, играющих чрезвычайно важную роль в нейрональных процессах.
Триптофан является незаменимой аминокислотой и не синтезируется в мозге высших животных. В мозге триптофан может переаминироваться с использованием щавелевоуксусной кислоты в качестве акцептора аминогруппы, а также декарбоксилироваться. Физиологическое значение первой реакции неизвестно. Наиболее интересный нейрональный путь метаболизма триптофана, которой составляет всего 5% от общего метаболизма триптофана в организме - это образование серотонина и мелатонина.
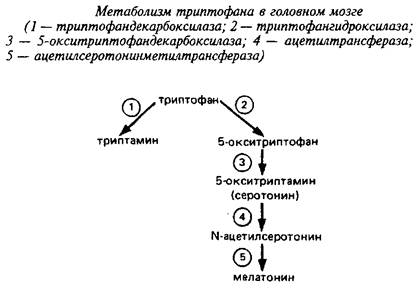
Первая ступень этого процесса - гидроксилирование триптофана в 5-м положении - катализируется триптофан-5-гидро-ксилазой. Энзим требует молекулярного кислорода и тетрагидробиоптерина в качестве кофактора. Этот фермент локализован исключительно в серотонинергических нейронах мозга. Он не полностью насыщен своим субстратом в мозге, Км для триптофангидроксилазы заднего мозга - 50 мкМ, а содержание триптофана там - 30 мкМ. Поэтому даже физиологические вариации уровня триптофана мозга влияют на синтез серотонина, а нагрузки триптофаном изменяют поведенческие реакции животных. Катехоламины являются сильными ингибиторами энзима, что говорит о тесной взаимосвязи между катехольными и индольными путями образования биоаминов.
Вторая ступень катализируется 5-окситриптофандекарбокси-лазой и ведет к образованию серотонина. В эпифизе серотонин при участии специфической ацетилтрансфера-зы ацетилируется с образованием N-ацетилсеротонина; последний подвергается О-метилированию с участием фермента метилтрансферазы, используя в качестве донора метильной группы S аденозилметионин При этом образуется гормон эпифиза мелатонин. Активность двух последних ферментов ответственна за изменение светового - темнового цикла у животных и зависит от циркадного ритма.
На содержание триптофана, а следовательно, и серотонина в мозге оказывает влияние характер используемой пищи; оно возрастает при приеме полноценных белков и богатой углеводами пищи. Углеводы стимулируют освобождение инсулина, который способствует поступлению в мышцы, а следовательно, удалению из циркуляции разветвленных аминокислот - конкурентов ароматических аминокислот за транспортные системы ГЭБ мозга. Таким образом, снижение уровня разветвленных аминокислот в плазме крови приводит к повышению транспорта ароматических аминокислот в мозг. Влияние пищи на поведение людей многие исследователи связывают отчасти с изменением уровня ароматических аминокислот в мозге, а отсюда и уровня биогенных аминов.
Запасы триптофана у животных составляют лишь 2-4% от дневной нормы, поэтому небольшие различия в скорости синтеза и катаболизма белка в зависимости от диеты или гормонального состояния могут вызвать большие изменения в уровне свободного триптофана. В регуляции уровня триптофана, а следовательно, и серотонина в мозге большую роль играет также кинурениновый путь катаболизма триптофана, реализующийся в печени. Этот путь инициируется триптофанпирролазой - печеночным ферментом, который использует главным образом триптофан из пищи и индуцируется как своим субстратом триптофаном, так и глюкокортикоидами. Гормон роста, напротив, предотвращает индукцию триптофанпирролазы триптофаном. Таким образом, триптофанпирролаза печени способствует удалению избытка триптофана из плазмы крови, что, в свою очередь, минимизирует изменение содержания триптофана в мозге.
Фенил аланин также является незаменимой аминокислотой и не синтезируется в мозге высших животных. В мозге происходит трансаминирование и декарбоксилирование фенилаланина. Эти реакции катализируются N-тирозин-2-оксоглутаратаминотранс-феразой и ДОФА-декарбоксилазой.
Главный путь метаболизма фенилаланина в целом организме - его гидроксилирование до тирозина с участием фермента фенилалашш-4-гидроксилазы - не обнаружен в мозге. Другие энзимы, присутствующие в мозге, могут катализировать гидроксилирование лишь небольшой части фенилаланина. Печеночная система гидроксилирования фенилаланина тщательно изучена, так как ее нарушение ведет к самому распространенному и тяжелому заболеванию, связанному с метаболизмом аминокислот, - фенилкетонурии. Система состоит из самой гидроксилазы, неконъюгированного птеридинового кофактора и пиридинсвязанной редуктазы для рециклизирования птеридинового кофактора. Гидроксилаза - сложный железосодержащий белок - является классической монооксигена-зой, требующей молекулярного кислорода в качестве окислителя, и L-эритротетрагидробиоптерина в качестве восстановителя. Второй энзим системы - дегидроптеринредуктаза - катализирует рециклизацию окисленного кофактора, используя НАДФН как источник электронов.
Фенилкетонурия - это следствие генетически обусловленного отсутствия фенилгидроксилазной активности в печени, она проявляется аминоацидурией с нарушениями нервной системы. Уровень фенилаланина в крови таких больных возрастает в несколько сотен раз по сравнению с нормой.
Заболевание сопровождается серьезными неврологическими нарушениями, включая конвульсии, тремор, умственные дефекты, необратимое и глубокое недоразвитие. Большинство больных детей гиперактивны и агрессивны, многие из них являются микроцефалами со слабой пигментацией кожи, волос, глаз. Из биохимических нарушений характерным является генерализованный дефицит миелина, сопровождающийся снижением уровней холестерина, цереброзидов, изменением отношения насыщенных и ненасыщенных жирных кислот.
Стандартная терапия фенилкетонурии - снижение доли фенилаланина в диете.
Тирозин - один из важнейших источников нейромедиаторов - катехоламинов. Превращение тирозина в катехоламины является главным путем метаболизма тирозина в мозге и надпочечниках. Первая ступень, катализируемая тирозин-З-гидроксилазой, лимитирует скорость всего процесса. Энзим является оксидазой со смешанными функциями, требующей кислорода, восстановленного птеридина и Fe+. Под действием фермента тирозин превращается в 3,4-дигидроксифени-лаланин. Активность его подавляется катехоламинами. Естественно, этот энзим служит надежным цитохимическим маркером нейронов, способных синтезировать катехоламины.
Декарбоксилирование ДОФА до дофамина выполняется ДОФА-декарбоксилазой, которая требует в качестве кофактора пиридоксальфосфат. В мозге энзим неспецифичен, действует на широкий спектр ароматических аминокислот, включая 5-гидрокситригттофан.
Дофамин-р-гидроксилаза, необходимая для его превращения в норадреналин, также присутствует в мозге. Показана необходимость молекулярного кислорода и аскорбиновой кислоты для его действия. Энзим содержит ионы меди и стимулируется фумаровой кислотой. Он неспецифичен и катализирует гидроксилирование боковых цепей большого количества р-фенилэтиламинпроизводных; в частности, тирамин является лучшим субстратом для энзима, чем дофамин.
Основной путь деградации тирозина в организме млекопитающих - через р-гидроксифенилпируват, гомогентизиновую кислоту и расщепление кольца - не встречается в головном мозге. В мозге присутствует Ь-тирозин-2-оксоглутаратами-нотрансфераза, которая осуществляет активное переаминирование тирозина в нейрональной ткани. Тирозин мозга является также субстратом для неспецифической декарбокси-лазы ароматических аминокислот.
Хотя известен ряд нарушений во всех путях деградации тирозина, ни один из них не вызывает тяжелых неврологических повреждений.
Гистидин не синтезируется в головном мозге, но он активно транспортируется через гематоэнцефалический барьер. В мозге гистидин может декарбоксилироваться, образуя гистамин - важный нейромедиатор и нейромодулятор. Декарбоксилирование гистидина могут выполнять два энзима. Первый из них - специфическая гистидин-декарбоксилаза, энзим, требующий пиридоксальфосфат в качестве кофактора. Он очень активен в ряде периферических нервов и в симпатических ганглиях. В то же время в головном и спинном мозге активность его мала; К^ для гистидина в нормальных условиях - порядка 410~ М. Фермент индуцируется при стрессе.
Вторым энзимом, который осуществляет декарбоксилирование гистидина, является неспецифическая декарбоксилаза ароматических аминокислот, действующая на ДОФА, 5-гидрокситриптофан, а также на гистидин. Представлено большое количество доказательств, что именно этот энзим является ответственным за декарбоксилирование гистидина в ЦНС.
Разрушение гистамина в целом организме происходит в основном при участии гистаминазы, но этот фермент отсутствует в мозге. Главный путь катаболизма гистамина в мозге - метилирование в 4-м положении с использованием S-аденозилметионина в качестве донора метальной группы при участии специфического энзима гистамин-метилтрансферазы. При подавлении этого энзима уровень гистамина в мозге сильно возрастает. Образованный метилгистамин затем окисляется до соответствующего альдегида и до метилимидазо-луксусной кислоты, которая экскретируется.
Концентрация гистамина в головном и спинном мозге низка, но он присутствует в значительных количествах в некоторых постганглионарных нервах. Его концентрация велика в переднем гипофизе и гипоталамусе. Субклеточно гистамин локализован преимущественно в синаптосомах.
10. Основные аминокислотыЛизин пока мало исследован в аспекте его значения для нервной системы. Пути деградации лизина в мозге точно не установлены, но они отличаются от локализованных в печени. Лизин в мозге может катаболировать через образование пипеколовой кислоты.
Интересно и важно, что нервная система исключительно чувствительна к нарушению метаболизма лизина в других тканях. Последнее приводит к тяжелым деструктивным и демиелинизационным процессам в ЦНС, сопровождающимся умственной отсталостью.
Аргинин в целом организме ассоциируется прежде всего с процессом синтеза мочевины. Однако в головном мозге не существует полного цикла образования мочевины, хотя некоторые энзимы этого метаболического пути, такие как аргинино-сукцинатсинтетаза, аргининосукдиназа и аргиназа, найдены в этом органе. Центральный фермент цикла - орнитинкарбамоилтрансфераза - не обнаружена в мозге.
Недавно выявлена еще одна важная функция аргинина. Он является источником образования окиси азота - мощного сосудорасширяющего фактора и нейромедиатора. Синтез N0 осуществляется с помощью фермента аргинат-синтазы. Образующийся при этом цитруллин включается в известный цикл образования мочевины.
Генетические дефекты, связанные с метаболизмом аргинина и образованием мочевины вне нервной ткани, сопровождаются неврологическими последствиями. Все эти генетические заболевания, такие как цитруллинемия, аргининосукцинатацидурия, аргининемия, сопровождаются накоплением в плазме крови и в тканях отдельных метаболитов аргинина. Но, вероятно, наиболее серьезным последствием таких метаболических блоков является сопутствующее им повышение концентрации ионов аммония - гипераммониемия, особенно опасная для растущего мозга и часто ведущая к коме. При аргининосукцинатацидурии умственная отсталость может быть очень тяжелой. Это заболевание сопровождается дегенеративными изменениями в белом веществе мозга, дефектами миелинизации и недоразвитием кортикальных слоев.
Метаболизм орнитина - диаминокислоты, являющейся ближайшим родственником аргинина, в нервной ткани открывает еще одну важную функцию аминокислот - они являются предшественниками полиаминов, соединений, которые выполняют пока мало изученный комплекс регуляторных функций.
Выводы
1. Аминокислоты широко используются для синтеза многих белков, пептидов, нейромедиаторов и других биологически важных соединений. Некоторые аминокислоты сами служат нейромедиаторами.
2. Состав пула свободных аминокислот в нормальных физиологических условиях отличается постоянством, отдельные районы мозга имеют свои характерные метаболические пулы.
3. Разнообразные активные транспортные процессы служат для поддержания уровней и распределения метаболитов как в целом органе, так и в отдельных его районах. Многообразие систем транспорта аминокислот ЦНС отражает полифункциональность этих соединений.
4. Пространственная разобщенность отдельных ступеней метаболизма аминокислот создает условия ддя пространственного разобщения энергетического метаболизма и не связанных с энергетикой функций и превращений аминокислот.
5. Головной мозг характеризуется высокой концентрацией аминокислот глутаминовой группы. Глутаминовая кислота, глутамин, ГАМК, аспарагиновая и N-ацетиласпарагиновая кислоты составляют в сумме 75% пула свободных аминокислот мозга.
6. Метаболизм аминокислот глутаминовой группы также чрезвычайно интенсивен. Эти аминокислоты выполняют ряд важных функций в ЦНС: энергетическую, служат для образования и устранения аммиака, выполняют роль нейромедиаторов и нейромодуляторов.
7. Ароматические аминокислоты имеют особое значение как предшественники катехоламинов и серотонина.
8. Нарушения, особенно генетические, в энзиматической системе метаболизма аминокислот часто имеют тяжелые неврологические последствия. Нарушение транспорта аминокислот в других органах часто также сопровождается неврологическими расстройствами.
Похожие работы
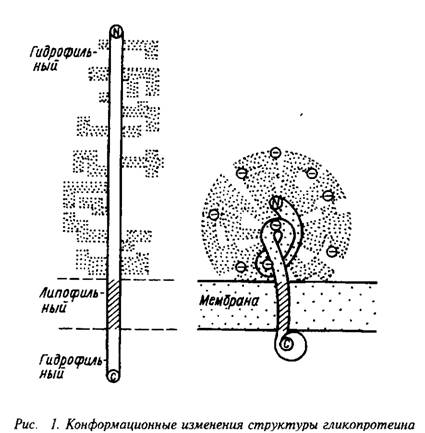
... , кроме того, в олигодендроцитах до миелинизации и в миелине периферической нервной системы. В ЦНС человека он представлен тремя полипептидными цепями с Мг=92, 107, 113 кД, а в периферической нервной системе – одним белком с Мг=107 кД. МАГ относится к гликопротеинам с относительно низким содержанием углеводных остатков – около 30% от массы молекулы, но содержит характерный для гликопротеинов набор ...
... транспорте. Гипокальциемия до 1,50-1,75 ммоль/л возможна в условиях хронической почечной недостаточности, гипопаратиреоза, авитаминоза Д. Сопровождается периферическими и центральными расстройства нервной системы. К периферическим нарушениям относятся парестезии и повышение возбудимости двигательных нервов. К центральным расстройствам относятся раздражительность, психозы, судороги, бред, ступор ...
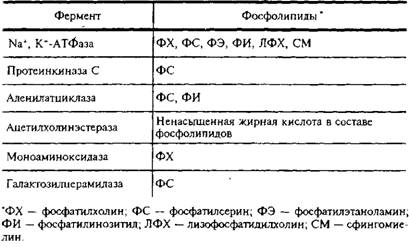
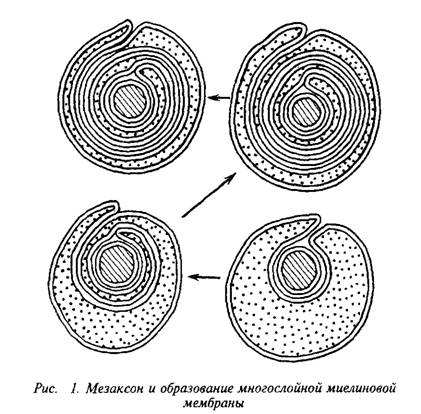
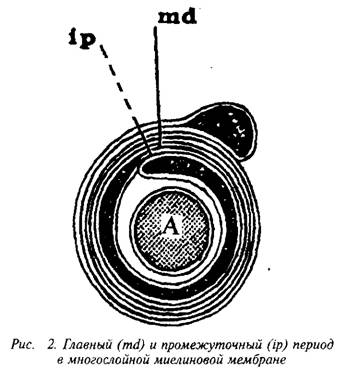
... и инозитолтрифосфат подвергаются химическим превращениям, требующим АТФ и ЦТФ и приводящим к восстановлению три-фосфоинозитида. Таким образом, цикл замыкается и уровень полифосфоинозитидов в мембране восстанавливается. 7. МИЕЛИН В ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЕ Мозг человека содержит 120 г миелина, что составляет одну треть его сухой массы. Миелин – уникальное образование, организация которого ...
... междисциплинарными связями. Студенты, приступающие к изучению нейрохимии, должны быть уже вооружены основательными знаниями по общей биохимии. Это обуславливает целесообразность преподавания нейрохимии на старшем курсе специальности «биохимия». Дисциплина относится к региональному (вузовскому компоненту). Распределение времени, отведенного на изучение дисциплины по учебному плану Форма ...
0 комментариев