КОНТАКТНОЕ ОКИСЛЕНИЕ И АДГЕЗИЯ К СТАЛИ ПОЛИЭТИЛЕНОВЫХ ПОКРЫТИЙ
Ранее [1,2] было установлено, что адгезия ПЭ-покрытий, окисляющихся на каталитически неактивной подложке, является функцией не только температуры Т и времени окисления т, но и толщины покрытий h. Уменьшение адгезии, характеризуемой удельным сопротивлением отслаиванию покрытий f при увеличении т обусловлено в основном проникновением к подложке низкомолекулярных продуктов термоокислительной деструкции макромолекул наружного поверхностного слоя покрытий. При повышении Т существенную роль в снижении величины f играет также уменьшение степени окисления граничащего с подложкой слоя полимера, связанное с ускорением процесса окисления в поверхностном слое покрытий.
Окисление граничащего с подложкой слоя осуществляется в основном кислородом, диффундирующим в покрытие из окружающей среды [3,4], а в катализе окисления полиолефинов на металлах существенную роль играют металлосодержащие продукты контактных реакций окисляющегося полимера с металлом, диффундирующие в покрытие (в основном соли карбоновых кислот) [5—7]. Поэтому следует ожидать, что закономерности изменения величины f для покрытий на каталитически активной подложке могут быть иными, чем на каталитически неактивной.
Цель данной работы — изучение адгезии ПЭ-покрытий толщиной 100—4000 мкм, окисленных на каталитически активной подложке (стали) [4,8,9] при различных температурно-временных условиях в среде воздуха: Г=433-453К, т=0,03-10,8 кс.
На рис. 1 представлены данные по влиянию т при различных температурах окисления на величину f для покрытий различной толщины на стали. Зависимость f от т при постоянных h и Т описывается кривой с максимумом. При увеличении h покрытий значение т, соответствующее максимуму f на изотермической кривой, сначала уменьшается, а затем увеличивается (рис. 1; рис. 2, б, кривая 4), в то время как для покрытий на каталитически неактивной подложке (алюминий [5]) оно только увеличивается [1]. Вследствие этого зависимость величины f покрытий на стали от h для постоянных Т и τ описывается при малых значениях τ кривой с максимумом, при средних т кривой с двумя максимумами, при больших т кривой с максимумом (рис. 3, а, кривые 1—3).
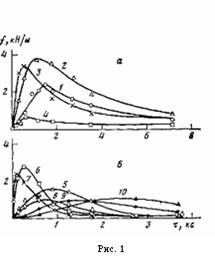
Рис. 1. Зависимость сопротивления отслаиванию ПЭ-покрытий на стали от времени их окисления т при 433 (а) и 453 К (б). Толщина покрытий 200 (1), 300 (5), 500 (2, 6), 800 (3, 7), 1000 (4, 8), 2000 (9) и 3000 мкм (10)
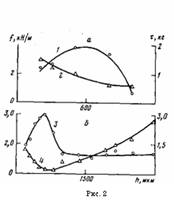
Рис. 2. Влияние толщины h ПЭ-покрытий на максимальные значения / (1, 3) для покрытий, окисленных при 433 (а) и 453 К (б), и время (2, 4), необходимое для достижения максимальной величины при 433 (а) и 453 К (б)
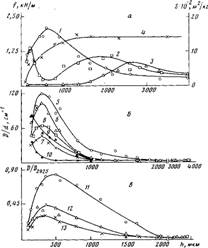
Рис. 3. Влияние толщины ПЭ-покрытий на стали, окисленных при 453 К в течение 0,6 (1), 1,2 (2, 10), 2,7 (S, 9) и 3,6 кс (3, 4, 5-7, 11-13), на сопротивление отслаиванию f (1—3), на площадь пика плавления S (4) (а), на накопление в граничащем со сталью слое солей карбоновых кислот (5, 8, 11), карбонильных (6, 9, 10, 12) и эфирных групп (7, 13) (б, в); б - данные, полученные методом ИК-спектроскопии пропускания для граничащего слоя толщиной 10 мкм; в — данные, полученные методом многократного нарушенного полного внутреннего отражения. Здесь и на рис. 5: D — оптическая плотность, d — эффективная толщина поглощающего слоя
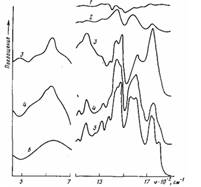
Рис. 4. ИК-спектры пропускания (1, 5) граничащего со сталью ело» толщиной 10 мкм (5) и остатков полимера на стали (1), а также ИК' спектры многократного нарушенного полного внутреннего отражения (2—4, 6) поверхности окисленной стали (6), стали, отслоенной от ПЭ-покрытия (2), и поверхности покрытия, контактировавшего со сталью, до (4) и после обработки в течение 7,2 кс раствором серной кислоты (3). Толщина покрытия 300 (1) и 500 мкм (2-5); время окисления при 453 К 0,6 (1); 1,8 (2); 3,6 кс (3-5)
При увеличении т для покрытий на стали в отличие от покрытий на алюминии f уменьшается после достижения максимума практически до нуля, а в слое, граничащем с подложкой, накапливаются соли карбоновых кислот — продукты контактных реакций карбоновых кислот, образующихся при окислении ПЭ с металлом. При понижении f с увеличением т уменьшается количество полимера, остающегося на подложке после ее отслаивания, и возрастает содержание на ней солей карбоновых кислот. При низких значениях f остающийся на подложке слой практически весь состоит из солей карбоновых кислот (полоса поглощения симметричных колебаний карбоксилат-аниона 1600 см-1, антисимметричных — 1440 см-1) (рис. 4, кривая 2). На отслоенной поверхности полимерного покрытия также фиксируются преимущественно соли карбоновых кислот (рис. 4, кривая 4). Следовательно, разрушение происходит по слою солей, а снижение f при увеличении т связано с их накоплением в зоне адгезионного контакта. Большое количество солей обнаруживается в слое толщиной 10 мкм, срезанном с поверхности отслоенного покрытия, бывшей в контакте с подложкой (рис. 4, кривая 5). Регистрируются они также в покрытиях, имеющих максимальные значения f (рис. 4, кривая 1). Кроме того, для отслоенного от подложки покрытия наблюдается сильное поглощение в области 500—700 см-1 (рис. 4, кривая 4), характерное для связи кислород — металл в карбоксилатах и оксидах железа (рис. 4, кривая 6). Оксиды железа в полимере могут образовываться вследствие окисления атомарного металла, появляющегося при распаде солей или при диффузии ионов и атомов металла в полимер. Ранее [1] было показано, что если к окисленному на каталитически неактивных подложках (алюминий, стекло) ПЭ-покрытию припрессовывать расплавленную неокисленную ПЭ-пленку, то значение f, снизившееся при увеличении τ, увеличивается вследствие перехода части низкомолекулярных продуктов окисления (карбоновых кислот и т. п.) из зоны адгезионного контакта в припрессованную пленку.
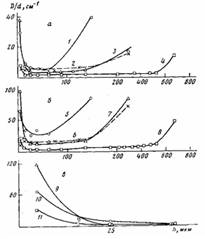
Рис. 5. Распределение по толщине h эфирных (а) и карбонильных групп (б), а также солей карбоновых кислот (в) для покрытий толщиной 200 (1, 5, 10), 300 (2, 3, 6, 7, 9) и 800 мкм (4, 8, 11), окисленных при 453 К в течение 3,6 кс на алюминии (2, 6) и на стали (1, 3, 4, 5, 7, 8, 9-11)
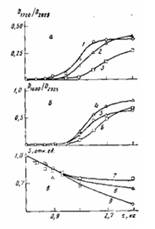
Рис. 6. Влияние времени окисления т при 453 К на накопление в граничащем со сталью слое ПЭ-покрытий толщиной 200 (1, 5, 9), 500 (2, 4, 8) и 800 мкм (3, 6, 7) карбонильных групп (а), солей карбоновых кислот (б) и изменение площади пика плавления S (в) на термограммах дифференциальной сканирующей калориметрии. Для зависимостей S—х толщина анализируемого слоя 10 мкм
Если проделать такую же операцию с покрытиями, окисленными на стали, то существенного восстановления значения f не происходит. Это также является свидетельством того, что причина спада величины f при окислении ПЭ-покрытий на стали заключается в накоплении в зоне адгезионного контакта малорастворимых в ПЭ солей карбоновых кислот.
Для условий, при которых значение f покрытий на стали с увеличением т падает (рис. 1, а), водостойкость адгезионных соединений увеличивается. Водостойкость повышается также при увеличении h покрытий, когда наблюдается возрастание концентрации солей в граничащем с подложкой слое. Однако такие адгезионные соединения ПЭ со сталью являются нестойкими в минеральных кислотах. Например, в водном растворе серной кислоты происходит самоотслаивание покрытий. Эти результаты легко объясняются, если учесть, что карбоксилаты железа нерастворимы в воде, но хорошо растворимы в серной кислоте и ее водных растворах. При растворении происходит реакция с образованием растворимых в воде сульфатов железа и карбоновой кислоты. Соли карбоновых кислот в отслоенных тонких покрытиях после их обработки в серной кислоте не обнаруживаются, а в толстых покрытиях их количество уменьшается (рис. 4, кривая 3). Как и следовало ожидать, одновременно увеличивается содержание карбоновых кислот в полимере, что характеризуется возрастанием интенсивности пика в области 1710—1720 см-1 (рис. 4, кривая 3). При этом уменьшается интенсивность поглощения в области 500—700 см-1. Последнее согласуется с предположением о том, что поглощение в указанной области связано с оксидами металла.
Карбоксилаты железа плохо растворимы в расплаве ПЭ и интенсивно накапливаются в граничащем с подложкой слое, при формировании Покрытий. Отметим, что аналогичные соли свинца, а также меди значительно лучше растворяются в расплаве ПЭ [5—7] и значение f для покрытий на свинце и на меди при увеличении т не уменьшается до нулевых значений [10,11]. При увеличении h толщина слоя покрытия, в котором регистрируются соли карбоновых кислот, уменьшается (рис. 5, в) вследствие локализации окисления в более тонком граничащем с подложкой слое покрытия (рис. 5, а, б, кривые 1, 3—8). Для ПЭ-покрытии на каталитически активной подложке в отличие от покрытий на каталитически неактивной подложке окисление наиболее интенсивно происходит в граничащем с подложкой слое, а также в поверхностном (наружном) слое (рис. 5, а, б). Для толстых покрытий окисление граничащего с подложкой и поверхностного слоев осуществляется независимо, а степень окисления граничащего с подложкой слоя меньше, чем у тонких покрытий (рис. 5, а, б, кривые 1, 4,5,8; рис. 6, а). При этом на определенной глубине от поверхности покрытия концентрация карбонильных групп резко уменьшается (рис. 5, б, кривая 8). По-видимому, на этой глубине концентрация кислорода в полимере недостаточна, чтобы преодолеть индукционный период окисления без каталитического воздействия подложки. Не исключено, что это связано с диффузией низкомолекулярных карбонилсодержащих продуктов окисления, образующихся в наружном слое. Для толстых покрытий, у которых окисление граничащего с подложкой и поверхностного слоев происходит независимо, растворимые в полимере низкомолекулярные продукты окисления, образующиеся в граничащем с подложкой слое полимера, могут диффундировать в среднюю неокисленную или малоокисленную часть покрытия. Эта часть покрытия для окисленного граничащего с подложкой слоя полимера может выполнять функцию «припрессованной пленки». При малых толщинах происходит взаимовлияние процессов окисления в граничащих с подложкой и в поверхностных слоях (рис. 5, а, б, кривые 1, 5). При этом концентрация карбонильных, эфирных и других кислородсодержащих групп в слое, граничащем с подложкой, уменьшается с уменьшением h покрытий при больших т (рис. 5, а, б; кривые 1,3,5,7), т. е. зависимость концентрации этих групп в граничащем с подложкой слое от толщины покрытия может описываться кривой с максимумом (рис. 3, б, в; кривые 6, 7,12,13; рис. 6, а). Аналогичной по характеру зависимостью описывается при этом накопление в граничащем с подложкой слое солей карбоновых кислот (рис. 3,6, в, кривые 5,8,11; рис. 6, б). Уменьшение концентрации кислородсодержащих групп и солей в граничащем с подложкой слое тонких покрытий на стали при уменьшении их толщины для постоянных Т и τ связано с участием каталитически активной подложки в окислении наружного поверхностного слоя покрытий. Соли карбоновых кислот лучше растворимы в частично окисленном ПЭ и регистрируются для более тонких покрытий, например, методом ИК-спектроскопии в более отдаленных от подложки слоях (рис. 5, в, кривая 10). При этом происходит более быстрое окисление поверхностного слоя (рис. 6, а, кривая 1), увеличивается окислительная сшивка макромолекул, способствующая аморфизации ПЭ, и раньше наступает характерный для окисления ПЭ-пленок на каталитически активных металлах (медь, свинец и др.) эффект автоингибирования окисления [5,12] (рис. 6, а, кривые 1,2). В результате уменьшается удельная (приходящаяся на единицу толщины), а также общая степень окисления покрытий (рис. 6, а, кривые 1,2; рис. 7). Чем выше степень окисления, тем больше образуется низкомолекулярных карбоновых кислот в покрытии. Следовательно, определенный вклад в накопление солей в граничащем с подложкой слое тонких покрытий могут вносить и карбоновые кислоты, диффундирующие из наружного слоя в зону адгезионного контакта. При окислении происходит аморфизация граничащего с подложкой слоя полимера (рис. 3, а, кривая 4; рис. 6, в). Если условия автоингибирования не достигнуты, то степень окисления ПЭ-покрытий, а также граничащего с подложкой слоя полимера уменьшается при увеличении h (рис. 3, б, кривые 9,10). Отметим, что основные изменения значения f происходят в условиях, когда автоингибирование еще не достигается (рис. 1, б; рис. 3; рис. 6, с).
Полученные данные показывают, что толщина покрытий существенно влияет на степень окисления граничащего с каталитически активной подложкой слоя, на накопление в нем солей карбоновых кислот и на адгезию покрытий к подложке. Для тонких покрытий при малых τ, когда автоингибирование окислительного процесса не достигнуто, увеличение толщины покрытия приводит к уменьшению скорости накопления карбонильных и эфирных групп в граничащем с подложкой слое (рис. 6, а, кривые 1,2; рис. 3, б, кривые 8,10) и к увеличению скорости накопления в нем солей(рис. 6, б, кривые 4,5; рис. 3, б, кривая 8). Так как спад величины f на зависимости f от τ обусловлен накоплением в зоне адгезионного контакта солей, смещение максимума f в область меньших т при увеличении h тонких покрытий связано с увеличением скорости накопления солей, а не с изменением степени окисления полимера, как можно было ожидать исходя из существующих представлений [8,9].
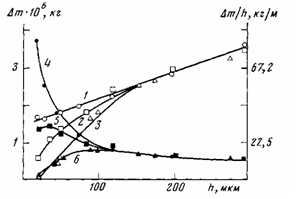
Рис. 7. Влияние толщины ПЭ-покрытий на алюминии (1—4), стали (2, 5) и меди (3, 6) на приращение массы Am (1—3) и удельное приращение массы Am/h (4-6), по данным ТТА, при окислении ПЭ-покрытий в иеизотермическях условиях
При этом зависимость максимальных значений f от h покрытий описывается кривой с максимумом (рис. 3, а, кривые 1,3), что можно объяснить уменьшением толщины окисленного граничащего с подложкой слоя полимера. Известно [13], что зависимость прочности соединений металлов с полимерами от толщины промежуточного тонкого окисленного полимерного слоя описывается кривой с максимумом. Определенную роль при этом может играть аморфизация граничащего с подложкой слоя полимера. Для толстых покрытий (1200 мкм и более) максимум величины f смещается в область больших значений τ при увеличении h (рис. 1, б, кривые 8—10), что связано, по нашему мнению, с влиянием толщины пленки на количество кислорода, проникающего в зону адгезионного контакта, т. е. с диффузионным ограничением контактного окисления.
Явления, аналогичные наблюдаемым при увеличении толщины ПЭ-пленок на стали (уменьшение т, соответствующего максимуму f, и увеличение при этом максимальных значений f [14], увеличение водостойкости [15]), наблюдаются при введении в ПЭ, контактирующий со сталью, неорганических наполнителей типа окиси алюминия, талька и др., относящихся к классу неорганических сорбентов. Ранее было показано, что эти наполнители способствуют локализации окисления, т. е. уменьшению толщины окисленной зоны ПЭ-пленок [16], препятствуют распространению зоны окисления [17], сорбируют карбоновые кислоты [18]. Это позволяет предположить, что в изменении адгезии ПЭ к стали при введении в него такого рода наполнителей существенную роль играют процессы локализации окисления, уменьшение в зоне адгезионного контакта карбоновых кислот и увеличение в этой зоне их солей.
ЛИТЕРАТУРА
1. Егоренков Н. Н., Кузаеков А. И. Высокомолек. соед. А, 1980, т. 22, № 11, с. 2498.
2. Егоренков И. И., Лин Д. Г., Кузаеков А. И. Докл. АН БССР, 1976, № 5, с. 417.
3. Белый В. А., Егоренков Н. И. Докл. АН БССР, 1970, № 12, с. 1100.
4. Лобанов Ю. Е. Механика полимеров, 1970, с. 932.
5. Егоренков Н. И., Лин Д. Г., Белый В. А. Докл. АН СССР, 1972, т. 207, № 2, с, 397.
6. Егоренков Н. И., Лин Д. Г., Белый В. А. Докл. АН СССР, 1974, т. 214, № 6, с. 1376.
7. Белый В. А., Егоренков Н. И., Лин Д. Г. Высокомолек. соед. Б, 1972, т. 14, № 10, с. 787.
8. Калнинь М. М. Автореф. дис. на соискание уч. ст. канд. хим. наук. Рига: Рижск. политехи, ин-т, 1967. 24 с.
9. Белый В. А., Егоренков Н. И., Плескачевский Ю. М. Адгезия полимеров к металлам. Минск: Наука и техника, 1971, с. 233.
10. Егоренков И. И., Лин Д. f., Белый В. А. Докл. АН СССР, 1973, т. 210, № 5, с. 1148. И. Егоренков Н. И., Лин Д. Г., Цыганок В. И., Белый В. А. Изв. АН БССР. Сер. физ.-техн. наук, 1974, № 2, с. 105.
11. Егоренков Н. И. В кн.: Металлополимерные материалы и изделия. М.: Химия, 1979, с. 19.
12. Песецкий С. С. Автореф. дис. на соискание уч. ст. канд. техн. наук. Рига: Рижск. политехи, ин-т, 1980. 16 с.
13. Калнинь М. М., Метниеце Е. О., Карливан В. П. Высокомолек. соед. А, 1971, т. 13, № 1, с. 38.
14. Калнинь М. М., Рейхманис П. К., Дзенис М. Я. Изв. АН ЛатвССР, 1980, № 2, с. 116.
15. Егоренков Н. И., Кузаеков А. И., Лин Д. Г. В кн.: Термический анализ. Рига: Зи-натне, 1979, т. 2, с. 7.
16. Егоренков Н. И., Белый В. А. В кн.: Надежность и долговечность полимерных материалов и изделий из них. М., 1969, с. 112.
17. Егоренков Н. И., Лин Д. Г., Кузаеков А. И. Высокомолек. соед. А, 1978, т. 20, № 8, с. 1385.
Похожие работы

... различных приборов и механизмов возникли новые требования в отношении свойств покрытий, в частности магнитных свойств. Эти требования в какой-то степени могут быть удовлетворены с помощью нанесения покрытий химическим способом из растворов, содержащих кобальт. Особое значение для звукозаписи и запоминающих устройств ЭВМ имеют тонкие магнитные пленки, которые получаются путем осаждения Со—Me на ...
... . После застывания парафина или церезина на поверхности предмета избыток его снимают, подогревая металлический инструмент и обтирая его мягкой тряпкой. Упаковывание является следующей стадией консервации. Консервация металлических изделий как средство временной защиты от коррозии на период транспортирования и хранения связана непосредственно с упаковкой, с 1971 г. введён в действие ГОСТ 13168-69 ...
... с кислородом, восстановлением - отнятие кислорода. С введением в химию электронных представлений понятие окислительно-восстановительных реакций было распространено на реакции, в которых кислород не участвует. В неорганической химии окислительно-восстановительные реакции (ОВР) формально могут рассматриваться как перемещение электронов от атома одного реагента (восстановителя) к атому другого ( ...
... Наименование источников информации, по которым проводился поиск Научно-техническая документация Патентная документация Способы изготовления полиэтиленовых труб Совершенствование технологии производства полиэтиленовых газопроводных труб для повышения качества продукции РФ МПК6 F16L 9/08 - 9/12 МПК7 B29D 23/00 Реферативный журнал «Химия. Технология полимерных материалов». ...
0 комментариев