М.М.Левицкий, В.В.Смирнов
Современное учение о катализе можно уподобить гигантскому живописному полотну, на котором с большого расстояния различимы два частично пересекающихся сюжета. Первый включает процессы, с помощью которых химики стремятся производить то, что давно умела делать природа. Речь идет в первую очередь о синтезе разнообразных органических веществ, получаемых живыми организмами буквально из земли, воды и воздуха. Не пытаясь воспроизвести природу, наши великолепные предшественники достигли многого, например, каталитически связали атмосферный азот. Современные успехи биохимии заставили по-иному взглянуть на многие процессы живого мира, где, как оказалось, властвуют истинные “короли” в мире катализаторов - ферменты. Они проводят синтезы в исключительно мягких условиях, с высокой селективностью и большим выходом.
Второй сюжет охватывает те процессы, которые природа делать не умеет, скорее всего за ненадобностью. Например, ни в живом, ни минеральном мире мы не встречаем полимеризации, галогенирования, нитрования и т.п. Тем не менее, реакции такого типа крайне необходимы для получения многих соединений, без которых современная цивилизация обойтись не может. О нескольких небольших зарисовках, дополняющих эту часть большой картины, пойдет речь.
Испытать металлорганосилоксаны
Модифицированием свойств натурального каучука химики занялись довольно давно. Один из самых удачных результатов - получение хлоропрена: в молекуле изопрена, который представляет собой мономер натурального каучука H2C=CH–C(CH3)=CH2, метильную группу заменили хлором - H2C=CH–C(Cl)=CH2.
Полимерный хлоропрен, называемый обычно хлоропреновым каучуком, исключительно устойчив к действию бензина и масел. Синтезируют хлоропрен в три стадии (рис.1). Сначала галогенируют бутадиен, в результате чего возникают два продукта - симметричный 1,4-дихлорбутен-2 (1,4-ДХБ) и несимметричный 3,4-дихлорбутен-1 (3,4-ДХБ). Затем отщепляют HCl от несимметричного соединения (симметричный продукт галогенирования не годится) действием щелочи и получают хлоропрен. Заканчивается синтез полимеризацией хлоропрена.
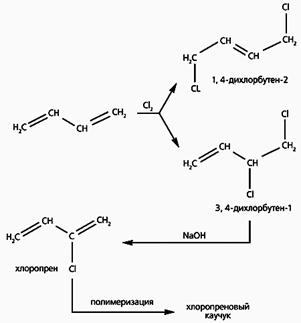
Рис.1. Схема получения полихлоропрена.
На двух последних стадиях выход очень высок - 95-98%, - и только на первой оставляет желать лучшего. Это связано с тем, что несимметричный продукт галогенирования, необходимый для дальнейших превращений, образуется в количестве почти вдвое меньшем, чем симметричное соединение. Чтобы увеличить выход конечного продукта, в технологическую цепочку включают еще одну стадию - каталитическую изомеризацию симметричного 1,4-дихлорбутена-2 в 3,4-дихлорбутен-1 (рис.2). Но применяемые для этого катализаторы - нафтенат меди либо ее галогенид - мало эффективны и довольно быстро теряют активность [1].
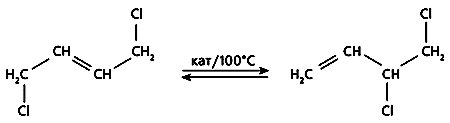
Рис.2. Изомеризация дихлорбутена.
В поисках новых катализаторов мы обратились к металлорганосилоксанам (МОС) - олигомерам (молекулярная масса 1500-2000), содержащим группировки –RSi–O–M–О–RSi–O–. По существу, МОС представляют собой металлосиликаты, которые обрамлены органическими группами [2]. Они эффективны в качестве катализаторов крекинга (Al, Zr-содержащие МОС), дегидратации спиртов (Mg-содержащие), галогенирования и селективного окисления углеводородов (Fe-содержащие). Каталитический центр любых металлорганосилоксанов - ион металла, находящийся в силоксановом окружении. Для изомеризации такие катализаторы ранее не применяли.
Нас они привлекли тем, что в процессе синтеза в их структуру можно вводить разные атомы металлов, плавно регулируя атомное соотношение M/Si. Кроме того, МОС растворимы в ацетоне, толуоле, спиртах, следовательно, пригодны как для гомогенного, так и гетерогенного катализа. Естественно, мы опробовали оба варианта.
Для исследования каталитической активности мы, казалось бы, должны следить за превращением симметричного продукта галогенирования (1,4-ДХБ) в целевое несимметричное соединение. Однако он для кинетических измерений не годится, потому что существует в виде цис- и транс-стереоизомеров, которые изомеризуются с несколько различающимися скоростями. В результате возникает необходимость следить за изменением концентрации двух веществ одновременно, что экспериментально сложнее и к тому же снижает точность измерений. Конечный продукт 3,4-ДХБ, в отличие от исходного, не образует стереоизомеров, мы этим воспользовались и стали измерять не то, что нужно, а то, что удобно.
Изомеризация - равновесная реакция (см. рис.2), следовательно, можно наблюдать не за прямым процессом, а за обратным, взяв в качестве исходного несимметричное соединение 3,4-ДХБ. Равновесие приведет в итоге к тому же соотношению компонентов реакции, что и прямой процесс. Катализатор не может сдвинуть равновесие: если в его присутствии увеличивается скорость прямой реакции, то возрастает и скорость обратной. Таким образом, задача катализатора - ускорить обе реакции, т.е. сократить время, которое необходимо для достижения равновесия. Далее задача решалась чисто технически. После того как достигалось равновесие, из реакционной смеси отгоняли несимметричный продукт (он кипит при более низкой температуре), а в остатке вновь восстанавливалась его равновесная концентрация в полном соответствии с законом действующих масс. Чем быстрее достигается равновесие, тем эффективнее процесс и, следовательно, тем лучше работает катализатор.
Выбранные нами металлорганосилоксаны, ранее не изучавшиеся в процессах такого типа, оправдали возлагаемые на них надежды.
Для гомогенного катализа были выбраны МОС с тем металлом, который используют в традиционных катализаторах, т.е. медь. Активность Cu-содержащих соединений с фенильной группой у кремния оказалась весьма высокой: конверсия в течение 80 мин достигала 75%, однако при большем времени работы металлорганосилоксан выпадал в осадок, образующийся из-за межмолекулярных сшивок [3]. В связи с этим мы стали ориентироваться на более устойчивые формы катализаторов, полученных нанесением МОС на поверхность силикагеля. Катализатор закреплялся на поверхности минерального носителя благодаря взаимодействию при повышенной температуре собственных остаточных гидроксильных групп с поверхностными гидроксильными группами силикагеля.
Перейдя к гетерогенному катализу, мы расширили набор металлов, вводимых в силоксановую цепь, и исследовали Cu-, Co- и Fe-содержащие органосилоксаны с фенильной группой у кремния [4].
Из экспериментов следовало, что наиболее активны Cu-содержащие фенилсилоксаны: конверсия 3,4-ДХБ в течение 60 мин составляла 72%, что почти в 4 раза превышало конверсию на эталонном катализаторе - аминном комплексе {[Cu·(NEt3)n]Cl2}m. Кобальт- и железосодержащие катализаторы проявили себя в ином: при изомеризации несимметричного 3,4-дихлорбутена возникал исключительно транс-изомер 1,4-дихлорбутена, в то время как на медьсодержащем катализаторе образовывалась смесь цис- и транс-изомеров. Полученные результаты интересны тем, что указывают путь к дальнейшему выяснению деталей механизма. Впрочем, повышенная каталитическая активность ионов меди в сравнении с остальными металлами удачно дополняет существующие представления о протекании процесса: работа катализатора связана с промежуточным частичным восстановлением металла. А среди исследованных нами металлов медь наиболее склонна к восстановлению.
Изучив подробнее изомеризацию при различном содержании Cu на носителе, мы установили, что процесс суммарно описывается кинетическим уравнением не совсем обычного вида: v = k[ДХБ][Cu]0.5. Показатель степени 0.5 указывает на то, что существенное увеличение количества катализатора не приводит к заметному возрастанию степени конверсии. Следовательно, его избыточное нанесение на силикагель нецелесообразно. Из этих результатов мы нашли, что оптимальное содержание каталитических центров фенилсилоксана на носителе составляет 0.6-0.8%.
Наиболее заметно новый катализатор отличается от изученных ранее исключительно высокой стабильностью каталитических свойств: многократно (6-10 раз) использованный, он почти не снижает активности.
Благодаря обнаруженным в экспериментах свойствам медьсодержащего фенилсилоксана мы смогли включить в исследования более широкий круг катализируемых реакций.
Галогенировать без элементарного галоида
Хлоруглеводороды служат исходными соединениями для синтеза самых разнообразных органических веществ. В химической промышленности чаще всего используется галогенирование элементарным хлором (заметим, такие производства загрязняют окружающую среду).
Существуют иные пути получения хлоруглеводородов, например, обменное галогенирование CCl4 с алканами (рис.3). Мы исследовали пару реагентов декан-CCl4, взаимодействие в которой катализировали комплексом галогенида меди [Cu·(ДМФА)n]Cl2. К сожалению, он быстро дезактивировался, и мы взялись за медьсодержащий фенилсилоксан. По каталитической активности он в 4-5 раз уступал комплексу меди, но отчетливо превосходил по стабильности каталитических свойств: его активность практически не снижалась после пятикратного использования, в то время как эталонный катализатор полностью терял работоспособность в течение одного цикла [5].
![]()
Рис.3. Обменное галогенирование CCl4 с углеводородом.
В ходе изучения процесса мы выяснили, что так же, как в рассмотренной только что изомеризации, увеличение количества катализатора не дает существенного выигрыша в конверсии. Чтобы детальнее исследовать это явление, мы оценили активность катализатора как конверсию, приходящуюся на один моль меди [6]. Оказалось, что максимальной активность бывает при весьма низком содержании каталитических центров на носителе - до 0.1%. Это нельзя было объяснить тем, что часть их находится в глубинных слоях катализатора и потому не участвует в катализе. Причины обнаружились при изучении особенностей строения самого катализатора.
В двух упомянутых катализаторах - комплексе галогенида меди и медьсодержащем фенилсилоксане - координационные сферы иона меди (т.е. атомы, окружающие Cu) различаются. Следовательно, сравнив работу этих катализаторов в обменном галогенировании, можно выяснить некоторые детали каталитического механизма.
A priori возможны три варианта:
- радикально-цепной механизм (с участием ионов меди переменной валентности);
- окислительное присоединение CCl4 к металлическому центру, протекающее в координационной сфере металла;
- стабилизация радикала ·CCl3 в координационной сфере металла.
Чтобы понять, какой именно вариант действует, мы исследовали поведение двух этих катализаторов при совместном хлорировании трех пар углеводородов [5]:
| декан–гексан | декан–толуол | гексан–толуол |
| С10Н22 – С6Н12 | С10Н22– С6Н5СН3 | С6Н12–С6Н5СН3 |
Два результата косвенно указывали на то, что реализуется радикально-цепной механизм: в толуоле хлорированию подвергалась преимущественно метильная группа; при хлорировании декана практически не затрагивались концевые группы СН3. Все это характерно для реакций отрыва атома водорода радикалом ·CCl3.
Мы рассудили, что если справедлива радикально-цепная схема, то при конкурирующем хлорировании двух углеводородов соотношение продуктов реакции должно соответствовать известным из литературы данным об активности атомов водорода при атаке связей С–Н радикалом ·CCl3. Кроме того, относительная реакционная способность не должна зависеть от типа комплекса, содержащего медь.
Если же процесс протекает в координационной сфере металла (как во втором и третьем вариантах), то особенность строения катализатора должна сказываться заметно, а соотношение продуктов должно отличаться от обычного соотношения активностей реакционных центров при атаке ·CCl3.
Результаты совместного хлорирования указанных трех пар углеводородов и поочередного использования двух катализаторов показали, что процесс протекает по радикально-цепному механизму. Действительно, соотношение продуктов хлорирования соответствовало взятым из литературы соотношениям реакционной способности атомов водорода при их отрыве радикалом ·CCl3 и практически не зависело от типа катализатора.
Обменное галогенирование мы изучили, используя еще один галогенирующий агент - CBr4 [7]. В итоге удалось выработать общую схему процесса (рис.4). Она состоит из двух равноправных ветвей. Первая - это превращение каталитического центра: согласно классической схеме, катализатор после участия в реакции возвращается в исходное состояние. Вторая ветвь - передача радикального центра по цепочке: от радикала ·CHal3 к углеводороду, затем вновь к CHal4 и т.д., в полном соответствии с определением радикально-цепных реакций.
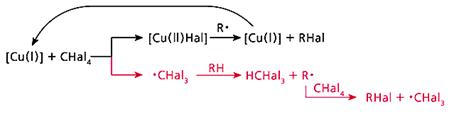
Рис. 4. Общая схема обменного галогенирования.
Верхняя ветвь - превращение каталитического центра, нижняя - радикально-цепной процесс.
В предложенной схеме есть одна деталь, которая требует пояснения. В медьсодержащем силоксановом катализаторе атом меди находится в состоянии Cu(II), а на схеме процесс начинается с Cu(I). Причину несоответствия удалось найти с помощью процедуры, которая позволяет обнаружить атомы меди, находящиеся в разной степени окисления. Исходные и выдержанные при 150°С (стадия, завершающая подготовку катализатора к дальнейшему его использованию) катализаторы мы обработали азотсодержащими лигандами и проанализировали их УФ-спектры [8]. Оказалось, что незначительная часть атомов меди при термообработке частично восстанавливается, и этого вполне достаточно, чтобы началась радикально-цепная реакция. Далее новые каталитические центры Сu(I) образуются благодаря возникающим в реакционной системе свободным радикалам.
Предотвратить координацию
В процессе обменного галогенирования хлор присоединяется к любому из вторичных атомов углерода в декане, не затрагиваются лишь концевые СН3-группы. В результате образуется смесь монохлордеканов. Такая скромная селективность обычно не удовлетворяет химиков; частично ее можно усилить заменив ненасыщенный углеводород насыщенным в качестве исходного реагента. Если вместо алкана взять алкен, положение двойной связи само укажет то место, куда должен присоединиться Cl, а также фрагмент CCl3 (рис.5).

Рис. 5. Каталитическое присоединение CCl4 к олефину.
Каталитическое обменное галогенирование в этом случае протекает тоже по радикальной схеме, но с некоторым отличием: при участии двойной связи олефина радикальный центр образуется легче. Факт вполне ожидаемый; радикальная полимеризация олефиновых мономеров именно поэтому столь широко распространена.
Так же, как в предыдущих двух процессах, катализатор сохранял стабильность каталитических свойств при многократном использовании [8]. Интересно, что вновь подтвердилась уже отмеченная особенность: катализатор проявляет максимальную активность (определяемую как конверсия, приходящаяся на один каталитический центр) при очень малом его содержании на носителе - до 0.2% меди. Очевидно, что прослеживается закономерность, и она требует объяснения.
Итак, большая часть атомов меди почему-то “выключена” из катализа. Нам удалось обнаружить причину этого с помощью магнитных измерений и спектральных исследований исходных металлорганосилоксанов. Выяснилось, что значительная часть атомов металла связана в межцепные координационные кластеры [2] (рис.6). Металл стремится заполнить свою координационную сферу, привлекая атомы кислорода соседних фрагментов –Si–O–M–, а оказавшись координационно-насыщенным, с трудом взаимодействует с реагентом CCl4.
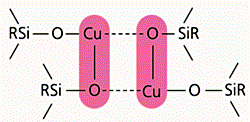
Рис. 6. Координационные межцепные кластеры в металлорганосилоксанах.
Как предотвратить образование межцепных кластеров? Можно, например, “укрыть” металл каким-либо лигандом. Это, безусловно, затормозит межцепную координацию, но не поможет решить основную задачу, поскольку лиганд в свою очередь затруднит приближение реагента. Тем не менее мы справились с ней, когда привлекли к участию органическую группу, связав ее с кремнием. Жесткая и мало подвижная фенильная группа, обрамляющая кремний в исследованных металлорганосилоксанах, не может препятствовать координационному взаимодействию атомов меди в соседних цепях. Если заменить фенильную группу на нонильную, ситуация меняется (рис.7). Объемистая алифатическая группа будто “окутывает” металлические центры и потому эффективно предотвращает координационное взаимодействие атомов меди в соседних цепях. Но в реакционной среде эта группа обладает подвижностью полимерного сегмента и не препятствует приближению органического реагента к каталитическому центру. Таким образом, четыреххлористый углерод как бы отодвигает нонильную группу, чего не может сделать атом меди соседней цепи.
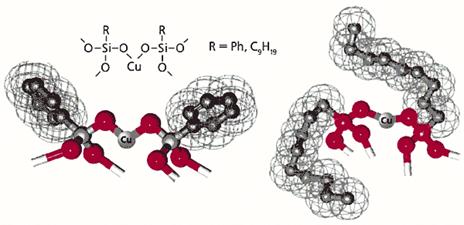
Рис. 7. Фенил (Ph)- и нонилсодержащие медьсилоксаны.
Сетчатая поверхность условно обозначает ван-дер-ваальсовы радиусы, т.е. ту часть пространства, которую действительно занимает фрагмент молекулы.
Судя по результатам экспериментов, в каталитическом присоединении CCl4 к октену-1 активность медьсилоксанов, обрамленных нонильными группами, вдвое выше, чем медьфенилсилоксанов [9].
Очевидно, что каталитические возможности металлорганосилоксанов не исчерпаны, впереди поиски новых способов структурного регулирования их каталитических свойств.
То живописное полотно, о котором говорилось в начале статьи, постоянно расширяется и дополняется. Но это не мешает нам смотреть с интересом на всю картину в целом, всматриваться в ее отдельные фрагменты, наблюдая за тем, как развивается замечательная область химии, именуемая катализом.
Список литературы
1. Смирнов В.В., Левицкий М.М., Невская С.М., Бучаченко А.Л. // Известия РАН. Сер. химическая. 1997. Т.1. С.209-210.
2. Левицкий М.М. // Российский хим. журн. 2002. Т.XLVI. №3. С.51-63.
3. Левицкий М.М., Кокорин А.И., Смирнов В.В. и др. // Известия РАН. Сер. химическая. 1998. Т.10. С.1946-1949.
4. Смирнов В.В., Левицкий М.М., Невская С.М., Голубева Е.Н. // Кинетика и катализ. 1999. Т.40. №1. С.86-89.
5. Смирнов В.В., Голубева Е.Н., Загорская О.А. и др. // Кинетика и катализ. 2000. Т.41. №3. С.439-442.
6. Смирнов В.В., Левицкий М.М., Тарханова И.Г. и др. // Кинетика и катализ. 2001. Т.42. №4. С.560-564.
7. Smirnov V.V., Zelikman V.M., Beletskaya I.P. et al. // Mendeleev Communication. 2000. №5. Р.175-176.
8. Смирнов В.В., Левицкий М.М., Тарханова И.Г. и др. // Кинетика и катализ. 2001. Т.42. №5. С.737-740.
9. Смирнов В.В., Левицкий М.М., Тарханова И.Г. и др. // Кинетика и катализ. 2003. Т.44. №4. С.625-628.
Похожие работы
... затрат в рециркуляционных реакционно-ректификационных процессах с различной организацией подачи рецикла для реакции изомеризации типа АВ. Глава 2. Расчетно-аналитическая часть 2.1. Анализ стационарных состояний рециркуляционного реакционно-ректификационного процесса. В рециркуляционных схемах существуют различные варианты подачи рецикла. В данном случае рассматривается схема, ...
... в клинической практике. Одним из таких препаратов является просидол. Изучаемый новый наркотический анальгетик просидол (производное оксипиперидина), разработан в Институте химических наук им. А.Б. Бектурова МН АН РК, под руководством члена-корреспондента АН РК д.х.н. профессора Пралиева К.Д. До начала проведения клинических исследований были получены данные от создателей препарата о его ...
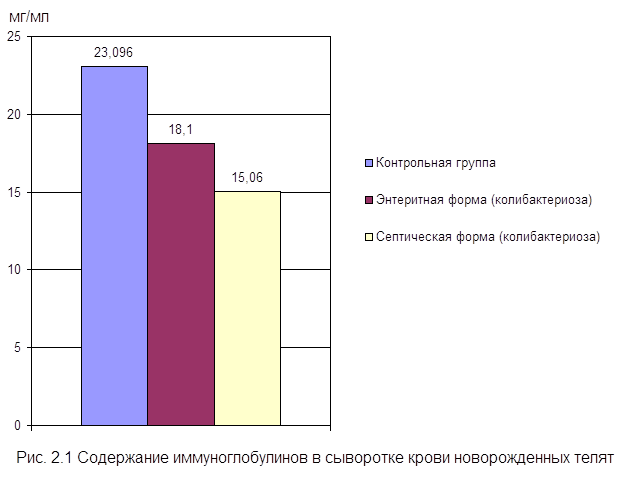
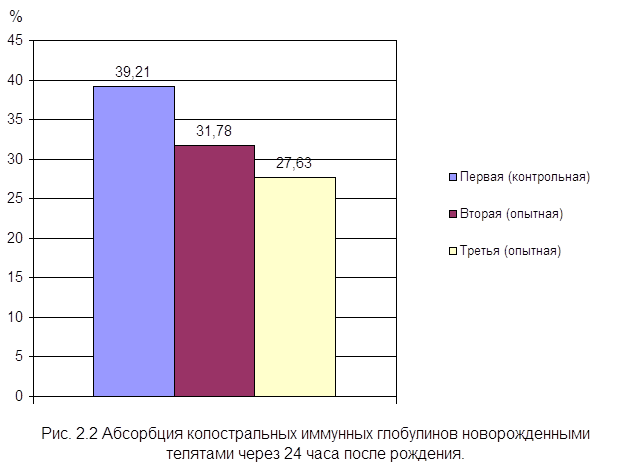
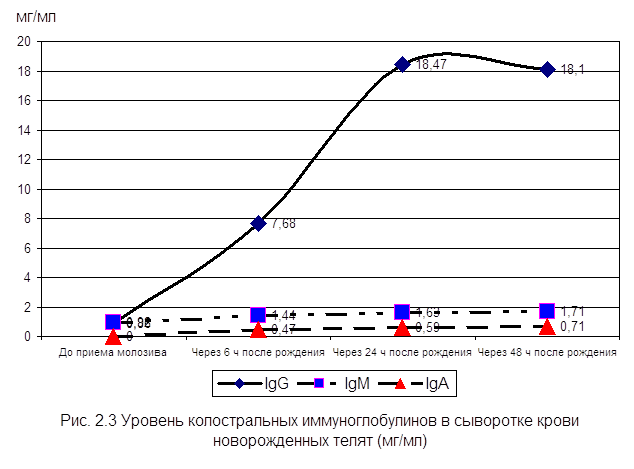
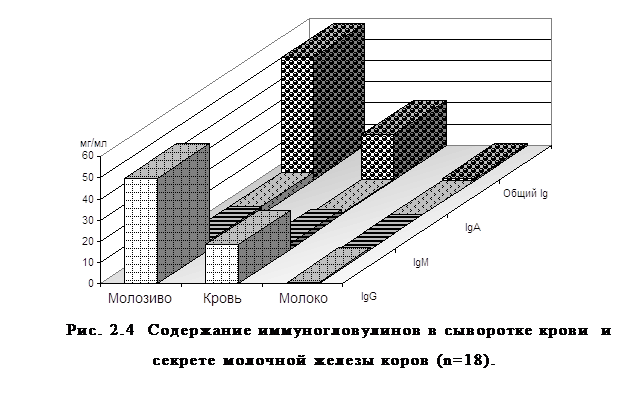
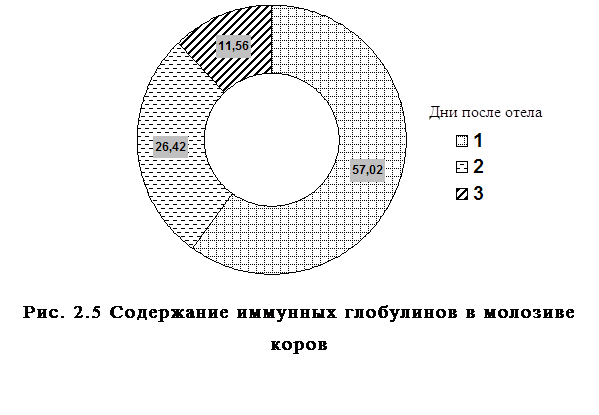
... , высокая эффективность послужит основанием для получения указанных препаратов в производственных условиях. заключение Анализируя данные литературы, касающиеся состояния естественной резистентности и иммунологической реактивности у новорожденных телят при колибактериозе, необходимо выделить следующие основные моменты: · колибактериоз занимает среди заболеваний новорожденных животных одно ...
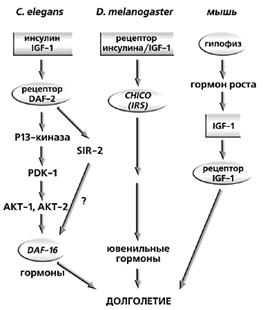
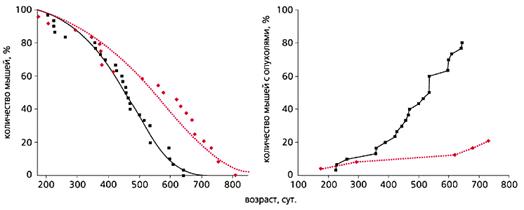
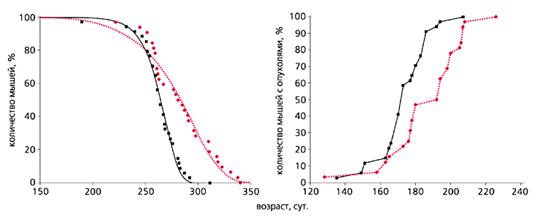
... мер против преждевременного старения и возрастной патологии. В короткой журнальной статье невозможно осветить даже бегло основные направления исследований современной фундаментальной геронтологии, поэтому отошлем читателя к соответствующей литературе [3, 4]. Мы коснемся лишь наиболее “горячих точек” в этой области, которая, по мнению журнала "Science", станет в ближайшие 25 лет одной из наиболее ...
0 комментариев