Навигация
Алкилирование енаминов, бета-дикетонов и енаминокетонов
28147
знаков
1
таблица
21
изображение
Новосибирский Государственный Университет
Кафедра Органической Химии
Курсовая работа по органической химии
студента II курса ФЕН Купрова И.С.
Тема: Алкилирование енаминов, β-дикетонов и β-енаминокетонов.
Научный руководитель:
д.х.н. проф. Ткачев А.В.
Новосибирск, 2000.
"The true worth of an experimenter
consists in his pursuing not only
what he seeks in his experiment,
but also what he did not seek."
Claude Bernard
Введение.
Классический подход к формированию связи углерод-углерод – реакция “нуклеофил плюс электрофил” – в настоящее время принимает самые разнообразные формы и является одним из основных инструментов синтетической органической химии. В качестве нуклеофилов в этих реакциях могут выступать разнообразные соединения, несущие неподеленную пару электронов, отрицательный заряд, либо активированную ароматическую систему. Во многих случаях нуклеофильность тесно соседствует с основностью – сродством к протону, которое может осложнять проведение нуклеофильных реакций за счет побочных процессов элиминирования.
В плане легкости образования карбаниона и одновременно высокого отношения нуклеофильность/основность из синтетически значимых нуклеофильных групп наибольшего внимания заслуживает атом углерода, находящийся на конце сопряженной системы, включающей в себя гетероатом (как правило, N либо O). Нуклеофильные свойства в этом случае обусловлены наличием неподеленной пары электронов и/или делокализованным отрицательным зарядом:
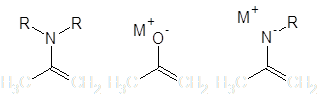
Соединения, содержащие подобные группировки широко используются как нуклеофилы в реакциях алкилирования. В этих структурах, однако, присутствуют два сравнимых по нуклеофильности центра – аллильный углерод и гетероатом, и большинство реакций в той или иной мере проходит по обоим этим положениям.
Введение в состав молекулы одновременно двух групп, стабилизирующих отрицательный заряд, существенно изменяет ее поведение в нуклеофильных реакциях, в частности, после подбора температуры и кислотности среды, оказывается возможным проведение селективного С-алкилирования. Интересным и синтетически значимым примером подобных соединений являются моноимины β-дикетонов, обычно существующие в термодинамически более стабильной енаминной форме:
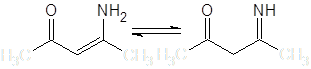
В настоящей работе сделан обзор литературных данных по реакциям С-алкилирования упомянутых групп соединений и исследована реакция бензилирования (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетона – енаминокетона, получаемого из природных терпеноидов.
Енамины, кетоны, β-дикетоны.
Синтезу и общим свойствам енаминов посвящена монография Дайка [1]. Как правило, енамины получаются взаимодействием карбонильных соединений с вторичными аминами (при получении с целью последующего алкилирования – обычно циклическими). В водных средах они гидролизуются, регенерируя исходные карбонильные соединения [2]:
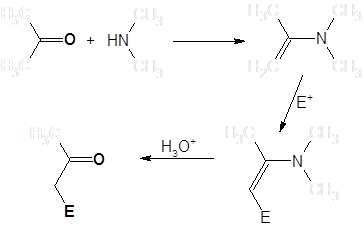
Скорости образования енаминов для различных вторичных аминов в большинстве случаев образуют следующий ряд:
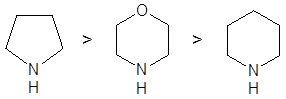
Условия протекания реакций алкилирования качественно одинаковы для всех рассматриваемых групп – щелочная среда или условия способствующие депротонированию α-углерода. Амминный атом водорода енаминов, также как Hα-С, в некоторой степени обладает кислотными свойствами, и в сильнощелочной среде реакция по гетероатому может конкурировать с основной. Особенно выделяются вторичные енамины, которые после N-депротонирования выступают как весьма активные N-нуклеофилы:
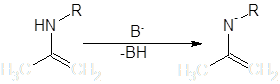
При их обработке электрофилом образуется от 10 до 70% N-замещенного продукта и многие авторы рассматривают C-алкилирование этих соединений лишь как побочную реакцию [3]. В целом, “семейство” побочных реакций для процессов нуклеофильного замещения довольно обширно, основные могут быть сведены в приведенную ниже схему (на примере енолятов) [4]:
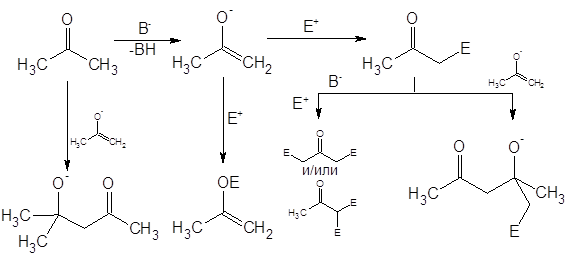
Авторы [4] приводят следующие условия как необходимые для протекания реакции алкилирования по требуемому направлению:
1. Скорость образования нуклеофильной частицы должна быть выше скорости ее конденсации с исходным веществом.
2. Селективное протекание реакции по атому углерода.
3. Скорость реакции взаимодействия нуклеофил-электрофил должна быть выше скорости реакции переноса протона.
4. Реакция нуклеофил-электрофил должна протекать быстрее, чем взаимодействие нуклеофила с продуктом реакции.
Из трех упомянутых в заголовке типов алкилируемых соединений безусловно лучшим или более удобным нельзя назвать ни один. Преимущества каждого описаны ниже:
| Енамин | Не требует высокоосновных/кислотных сред для образования или реакции. |
| Енолят (лития) | Легко и быстро генерируется, реагирует с предсказуемой региоселективностью. |
| Анион иминия | Исключительно низка скорость протонного переноса, предсказуемая региоселективность. |
Две основные проблемы, возникающие в реакциях алкилирования енаминов – полизамещение и реакции по атому азота, наиболее наглядно проявляют себя в случае алкилирования алкилгалогенидами. Это проиллюстрировано результатами настоящей работы и данными авторов [4] (метилирование), опубликовавших системные исследования по этой тематике:
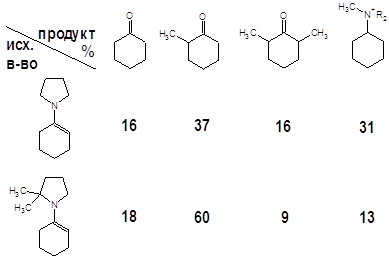
Вторая строчка таблицы несколько “лучше” – за счет наличия двух метильных групп реакция протекает более или менее однозначно. Стерические затруднения, характерные для более сложных кетонов, снижают долю полиалкилированых продуктов, хотя и увеличивают время реакции [4]. Те же пространственные факторы во многих кетонах ограничивают список потенциальных алкилирующих агентов следующими:
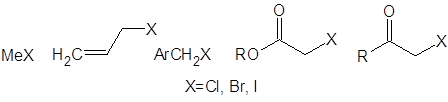
Даже с этими соединениями алкилирование кетонов через еноляты лития обеспечит высокий выход лишь для кетонов, устойчивых в сильноосновной среде (необходимой для генерирования енолятов). В случае β-дикетонов для депротонирования требуется менее основная среда. В работе [5] исследовано влияние пространственного строения бокового алкильного заместителя на скорость и региоселективность реакций алкилирования β-дикетонов:
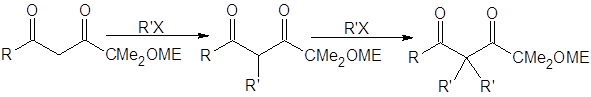
В противоположность алкилированию галогенидами, присоединение по Михаэлю активированных алкенов проходит с высокой специфичностью по атому углерода. Реакция по гетероатому в данном случае оказывается обратимой и сдвиг к термодинамически более стабильному продукту С-алкилирования приводит к хорошим выходам соответствующих алкилпроизводных:
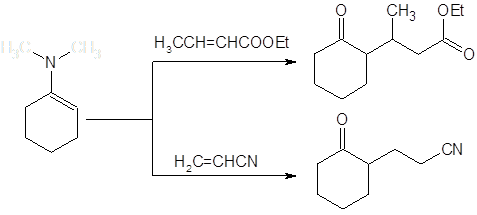
В отличие от кето-енольной таутомерии, факторы, контролирующие енамин-иминные превращения, изучены мало. Известно, что стабильность енаминных форм выше для третичных и ниже для первичных ненасыщенных аминов, но даже первичные и вторичные енамины могут быть стабилизированы введением в β-положение соответствующего заместителя (в работе [6] – COOH). Показано также, что полярные растворители сдвигают равновесие в сторону образования енаминов. Данные теоретических расчетов, в то же время, дают противоположный результат [6]. Для некоторых катион-радикалов енаминов и иминов и соответствующих нейтральных молекул измерены теплоты образования [7].
Енамин-иминная таутомерия может обуславливать быструю рацемизацию некоторых веществ в протонных растворителях, препятствуя тем самым разделению энантиомеров соединений, содержащих в углеродном скелете группировку
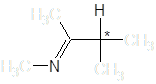
Авторы [8] наблюдали рацемизацию (S)-тетрагидрозолина, обусловленную таутомерными переходами:
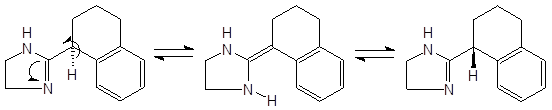
Исследование кинетики реакции рацемизации показало, что содержащие протон асимметрические центры по соседству с иминной группой “долго не живут”.
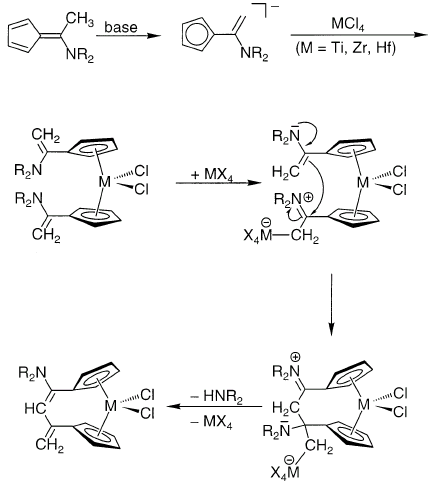
Реакции ароматических енаминов могут катализироваться тетрагалогенидами Ti, Zr, Hf [9]. В качестве любопытного примера реакции алкилирования енамина можно привести осуществленную совсем недавно межлигандную конденсацию [10]:
β-енаминокетоны.
Один из наиболее удобных синтетических методов получения сложных енаминонов, в том числе оптически активных – синтез на основе природных терпеноидов – лимонена, 3-карена и δ-кадинола. Эти соединения, интересные сами по себе (отмечена их биологическая активность), являются ключевыми промежуточными продуктами в асимметрическом синтезе [11]. Енаминокетоны, полученные из этих терпенов, используются как хиральные основания для разделения энантиомеров оптически активных кислот [12].
Енаминная система в щелочных средах может быть депротонирована и продукт введен в реакцию с алкилирующими агентами.
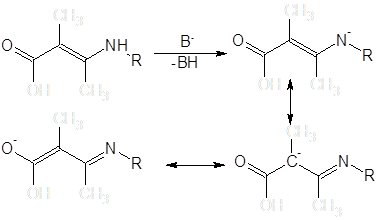
До последнего времени считалось, что преобладающими продуктами реакций алкилирования енаминокетонов являются N-замещенные производные [13]. Данные последующих исследований показывают, что алкилирование некоторых β-енаминокетонных систем в условиях межфазного переноса может быть селективно проведено и по атому углерода [14]:
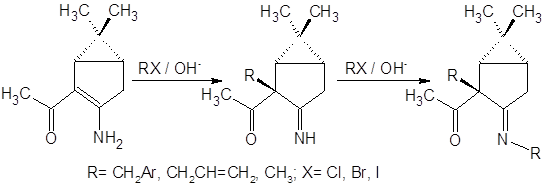
Приведенная последовательность превращений характерна, однако, только для алкилирования стерически нагруженными алкилгалогенидами. При использовании в качестве галогенида йодистого метила образуются все продукты вплоть до пентазамещенного. Исходя из данных настоящей работы, можно отметить, что направление алкилирования приведенных на рисунке соединений существенно зависит также от температуры реакции: при 35°С преобладает продукт С-алкилирования, при более низких температурах из смеси удается выделить N-алкилзамещенный продукт, доля С-замещения при этом невелика.
В работе [13] исследовано алкилирование 3-амино-5,5-диалкилциклогекс-2-ен-1-она для различных алкильных заместителей и проведен анализ факторов, необходимых для селективного направления алкилирования по тем или иным положениям изученного енаминона:
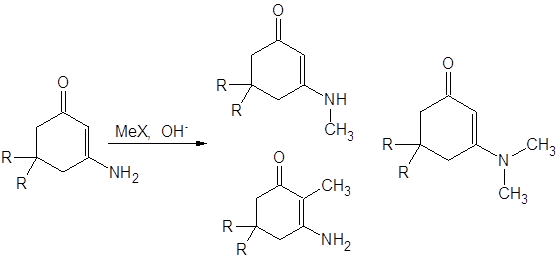 Зависимость хода реакции алкилирования аналогичного [13] циклического енаминокетона от природы боковых радикалов изучена авторами [3]:
Зависимость хода реакции алкилирования аналогичного [13] циклического енаминокетона от природы боковых радикалов изучена авторами [3]:
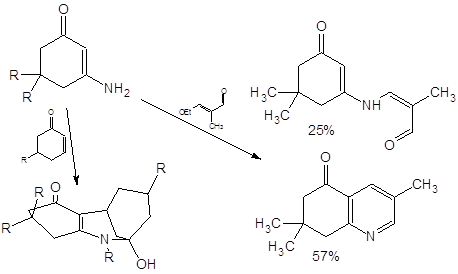
Среди известных реакций енаминонов внимания также заслуживает описанная в [15] реакция фотоарилирования:
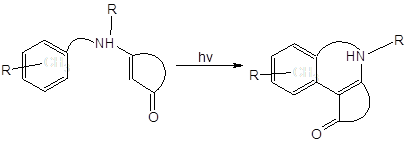
Авторы утверждают, что облучение енаминонов светом с длиной волны < 300 нм “can result in the formation of a variety of products… …photoreductions predominate”.
Из приведенных литературных данных можно сделать выводы об условиях, необходимых для получения высоких выходов С-алкилированных производных енаминов, b-дикетонов и енаминонов:
1. Необходим подбор основности среды. В низкоосновных средах мала концентрация активного аниона и реакция протекает медленно, в слишком высокоосновной среде происходит депротонирование атома азота и преобладающим становится продукт N-алкилирования.
0 комментариев