Навигация
Редкие причины нефротического синдрома
19185
знаков
0
таблиц
0
изображений
2. Редкие причины нефротического синдрома
Ишемическая болезнь почек
Нефротический синдром при ишемической болезни почек встречается редко. Обычно его относят к проявлениям гломерулонефрита, а стеноз почечных артерий не диагностируют. Применение малых доз ингибиторов АПФ или реваскуляризация нередко оказываются эффективными средствами борьбы с ним. Описаны случаи исчезновения нефротического синдрома при применении ингибиторов АПФ, нефрэкгомии ишемизированной почки, после реваскуляризации стенозированой почечной артерии. В большинстве случаев нефротического синдрома при сосудистых поражениях почек при гистоморфологическом исследовании выявляют фокально-сегментарный гломерулосклероз, являющийся в данном случае вторичным, а не самостоятельным заболеванием. Генез нефротического синдрома, вероятно, обусловлен выраженной гиперренинемией и гиперангиотензинемией, обладающими протеинурическим действием.
Нефротический синдром при гидатидной форме эхинококкоза
Описаны единичные случаи нефротического синдрома при эхинококкозе. При проведении пункционной нефробиопсии у пациентки 67 лет, страдающей мембранозным гломерулонефритом и эхинококкозом печени (гидатидной формой), в биопсийном материале были обнаружены антигены Есhinococcus granulosis. Наблюдали ребенка 4 лет, страдающего эхинококкозом легких и нефротическим синдромом, который был купирован при 8-недельной терапии албендазолом. Описан случай развития нефротического синдрома в рамках узелкового полиартериита, возникшего вследствие эхинококкоза. Описаны также случаи развития мембранопролиферативного гломерулонефрита, липоидного нефроза вследствие эхинококкоза печени (гидатидной формы) и нефротического синдрома вследствие эхинококкоза легких (гидатидной формы). Учитывая небольшое количество сообщений, представляется невозможной попытка выделения особенностей течения неф-ропатии при эхинококкозе. В настоящее время известно, что, как правило, встречается гломерулонефрит, в генезе которого лежит развитие аутоиммунной реакции с первоначальной ролью антигена эхинококка. Следует предположить, что лечение эхинококкоза с последующим наблюдением и лечением гломерулонефрита является основной тактикой ведения данной категории пациентов.
Геморрагическая лихорадка
Инфекционное заболевание, вызываемое вирусами семейства Вunyaviridae, рода Наntavirus. Подтипы гантавирусов Наntaat, Dobrova аnd Seoul являются причинами среднетяжелых и тяжелых форм геморрагической лихорадки с поражением почек в Азии и Европе, подтип Рuumala — легкой формы, встречающейся в Центральной Европе и Скандинавских странах. Для диагностики инфекции широко используется полимеразная цепная реакция с выделением РНК-вируса из крови и мочи. Имеются единичные наблюдения развития нефротического синдрома.
Болезнь Kimurа
Болезнь Кimurа представляет собой воспалительное заболевание неясной этиологии, распространенное в странах Азии (Китай, Япония). Страдают им в основном лица мужского пола. Клиника представлена триадой симптомов: подкожные болезненные массы в области головы и шеи, эозинофилия крови и тканей и повышенный уровень IgЕ в плазме крови. Болезненные массы являются увеличенными лимфатическими узлами, односторонние. Поражаются в основном периаурикулярные, аксиллярные и ингвинальные лимфоузлы.
При гистологическом исследовании биопсированного лимфатического узла выявляется фолликулярная лимфоидная гиперплазия с интранодальными и перинодальными эозинофильными инфильтратами и микроабсцессами. При иммуногистохимическом анализе выявляются антитела к СD20, СD3, СD45, СD15, СD30. В 12-16% случаев при болезни Kimurа наблюдается поражение почек, клинически проявляющееся протеинурией. Одним из наиболее частых синдромов является нефротический. При гистологическом исследовании выявляются липоидный нефроз, мезангиопролиферативный гломерулонефрит, мембранозный гломерулонефрит или фокально-сегментарный гломерулосклероз.
Нефротический синдром при situs viscerus inversus
Situs viscerus inversus встречается с частотой 1:5000 — 1:10 000 живорожденных. Domanski М. с соавт. (2005) описал случай нефротического синдрома у 18-летней девушки с situs viscerus inversus. При проведении нефробиопсии выявлен липоидный нефроз. Не исключено, что сочетание этих двух патологических состояний является случайным.
Диффузный фиброзирующий альвеолит (синдром Хаммена-Рича)
Редким проявлением болезни может быть поражение почек с развитием нефротического синдрома. Встречается при идиопатическом фиброзирующем альвеолите, а также при вторичном, развивающемся вследствие системных заболеваний соединительной ткани.
Галактосиалидоз
Галактосиалидоз является редким заболеванием, обусловленным мутацией гена протективного белка, локализованного на длинном плече 20-й хромосомы, наследующимся по аутосомно-рецессивному типу и характеризующимся дефицитом лизосомальных ферментов — а-нейраминидазы (сиалидазы) и Р-галактозидазы (Аксенова М.Е. и соавт., 2005).
Больные имеют типичный для болезни накопления Гурлерподобный фенотип, висцеромегалию, неспецифические диффузные изменения скелета (дизостоз), нередко тугоухость и изменения на глазном дне по типу «вишневой косточки». Гурлерподобный фенотип включает в себя низкий рост, непропорционально большую голову, грубые черты лица, уплощенную переносицу, короткую шею, вальгусную деформацию нижних конечностей, плоскостопие, пупочную и пахово-мошоночную грыжи. Поражение почек проявляется нефротическим синдромом (Аксенова М.Е. и соавт., 2005). В постановке диагноза помогает сочетание нефротического синдрома с Гурлерподобным синдромом. Для точной верификации диагноза применяется определение в крови а-нейраминидазы и Р-галактозидазы, резко сниженных. Однако данный вид анализа не является распространенным и применяется в крупных педиатрических центрах. При инфантильном типе гибель наступает во внутриутробном периоде или вскоре после рождения. При ювенильном типе болезнь проявляется в детстве.
При нефробиопсии обнаруживают накопление сиалоолигосахаридов высокой молекулярной массы преимущественно в подоцитах и клетках проксимальных канальцев, очаговое расширение мезангиальных площадок в гломерулах, фокальный гломерулосклероз.
Болезнь Кастельмана
Болезнь Кастельмана представляет собой гигантскую или ангиофолликулярную гиперплазию лимфатических узлов. Описано два типа поражения лимфатических узлов: гиалинваскулярный и плазмаклеточный. При гиалинваскулярном типе, встречающемся в 80-90% случаев, наблюдается пролиферация капилляров герменативных центров лимфатических фолликулов с их гиалинизацией. Классический вариант течения характеризуется поражением медиастинальных лимфатических узлов. В 20-30% случаев встречается плазмаклеточный тип с гиперплазией лимфатических фолликулов и инфильтрацией их плазматическими клетками. Описан также смешанный тип. Редким является генерализованное течение болезни с поражением шейных, абдоминальных и медиастинальных лимфатических узлов. Описаны случаи сочетания генерализованной формы болезни Кастельмана с нефротическим синдромом. При гистологическом исследовании диагностируется АА-амилоидоз почек, мезангиопролиферативный гломерулонефрит.
3. Семейные и врожденные формы нефротического синдрома
Семейный идиопатический нефротический синдром
Семейный идиопатический нефротический синдром развивается в детском возрасте и наследуется чаще по аутосомно-рецессивному типу или у взрослых и наследуется по аутосомно-доминантному типу. При семейном нефротическом синдроме с аутосомно-рецессивным типом наследования выявлен ген КРН82, локализующийся в 25-м локусе 1-й хромосомы и отвечающий за стероидорезистентность. Как правило, морфологическим эквивалентом является фокально-сегментарный гломерулосклероз. Ген кодирует синтез гломерулярного белка подоцина. Развивается в детском возрасте и характеризуется низкой протеинурией в дебюте с последующим развитием нефротического синдрома и постепенным прогрессированием ХПН. Отмечается резистентность к терапии глюкокортикостероидами.
В развитии семейного нефротического синдрома с аутосомно-доминантным типом наследования играют роль мутации генов, локализующихся в 13-м локусе 19-й хромосомы (FSGS1) и 21-22-м локусах 11-й хромосомы (FSGS2). FSGS1 приводит к формированию дефекта а-актинина-4. При мутации FSGS1 и FSGS2 наблюдается появление протеинурии в подростковом возрасте и сравнительно медленное прогрессирование с появлением спустя 3-5 лет нефротического синдрома. Исходом является ХПН.
Врожденный нефротический синдром финского типа
Распространен в Финляндии и встречается с частотой 1:8200 новорожденных. Ген (NPHS1), мутация которого является причиной развития данной патологии, локализуется в 13-м локусе 19-й хромосомы. Известно четыре гаплотипа, наблюдающихся у 90% семей с финским типом нефротического синдрома, что позволяет производить пренатальную диагностику в семьях больных. Ген NPHS1 кодирует синтез нефрина. Мутация гена NPHS1 обнаруживается и при других наследственных формах нефротического синдрома. Описано порядка 36 различных мутаций этого гена.
Характерным клиническим синдромом является нефротический, появляющийся на первой неделе жизни. Вес плаценты превышает вес ребенка при рождении более чем на 25%. Наблюдается резистентность к терапии глюкокортикостероидами и цитостатиками. Пациенты с Fin-minor-типом мутации рефрактерны к терапии ингибиторами ангиотензинпревращающего фермента, вместе с тем тип Fin-maior имеет более доброкачественное течение. Эффективным методом терапии является почечнозаместительная терапия.
Литература
1. Нефрология. Ключи к трудному диагнозу/ М.М.Батюшин – Элиста: ЗАОр НПП «Джанагар», 2007.
2. Нефрология. Основы диагностики / Под ред. дроф. В. П. Терентьева. (Серия «Медицина для Вас».) — Ростов н/Д: Феникс, 2003.
Похожие работы
... сыпь имеет сходную гистологическую характеристику с таковой при семейном холодовом аутовоспалительном синдроме. При проведении дифференциальной диагностики следует обратить внимание на лихорадку, болевой синдром в животе и суставах, не характерные для нефротического синдрома, обусловленного хроническим или быстропрогрессирующим гломерулонефритом. Нефротический синдром, ассоциированный с WT1- ...
... В клинической практике наиболее часто встречаются такие причины развития нефротического синдрома, как диабетическая, паранеопластическая нефропатия и хронический гломерулонефрит. Для дифференциальной диагностики причин нефротического синдрома требуется в первую очередь информация, полученная при беседе с больным и анализе имеющейся медицинской документации, а также при объективном обследовании. ...
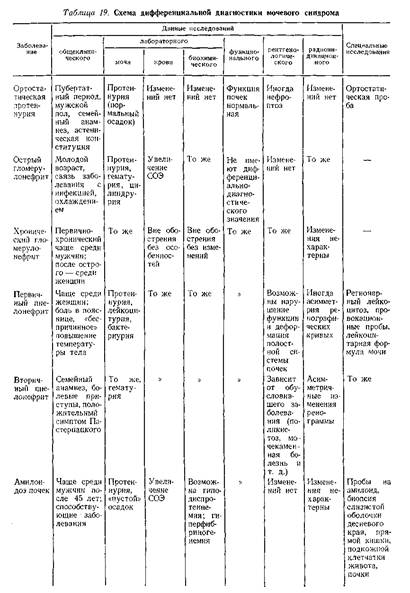
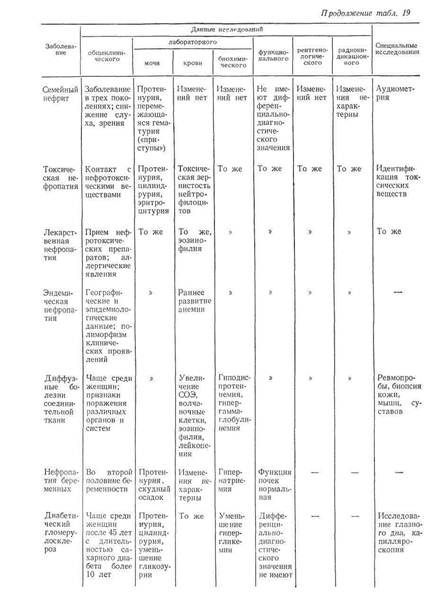
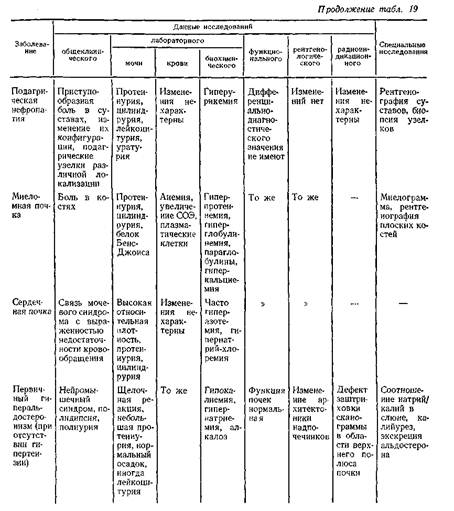
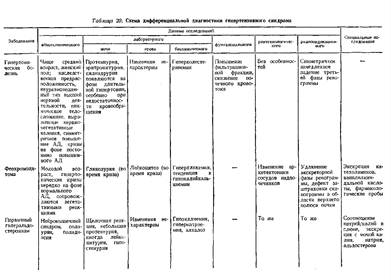
... заболеваниями соединительной ткани приходится с гломерулонефритом. О наличии сердечной почки свидетельствует положительная динамика мочевого синдрома в связи с уменьшением недостаточности кровообращения. Возможно сочетание гломерулонефрита и сердечной почки. ГИПЕРТЕНЗИВНЫЙ СИНДРОМ ПРИ ЗАБОЛЕВАНИЯХ ПОЧЕК Артериальная гипертензия является одним из самых частых симптомов при заболеваниях почек. ...
... в качестве причины нефротического синдрома заподозрена гипернефрома (наличие выраженной гематурии, прощупываемой опухоли, метастазов и других симптомов), должно быть проведено тщательное урологическое исследование. Нефротический синдром встречается в 10–30% случаев системной красной волчанки и чаще наблюдается у женщин молодого возраста. Иногда он может быть начальным проявлением волчанки. В этом ...
0 комментариев