Показания для назначения стабилизаторов настроения
Блокаторы кальциевых каналов, верапамил (финоптин, изоптин), нифедипин (адалат, коринфар) и дилтиазем (кардизем)
Пантогам. Кальциевая соль D-(+)-a,y, диокси-b-b-диметилбутирил-аминомаслянаякислота
Гидрохлорид b-диметиламиноэтилового эфира п-хлорфеноксиуксусной кислоты (IV)
Пикамилон
Количественное определение
Фотометрический метод определения ацефена [21]
Навигация
Гидрохлорид b-диметиламиноэтилового эфира п-хлорфеноксиуксусной кислоты (IV)
Ноотропные и нормотимические препараты
106341
знак
5
таблиц
12
изображений
2. Гидрохлорид b-диметиламиноэтилового эфира п-хлорфеноксиуксусной кислоты (IV)
К раствору основания (4) в хлорбензоле, нагретому до 50°, прибавляют 250 мл 18-22% раствора хлористого водорода в безводном спирте (по конго). Смесь оставляют на 20 часов при температуре 2-3°, кристаллы (4) отфильтровывают и промывают дихлорэтаном. Получают 0,3 кг (4), т. пл. 133‑135°, которые перекристаллизовывают из 3 л безводного дихлорэтана. Выход (4) 0,25 кг (50% на I), т. пл. 156-158°.
Фармакопейный ацефен может быть также получен при перекристаллизации технического (4) из спирта (1:1) [14].
5.2 Карбамазепин
10,11-Дигидро-5-хлоркарбонил-5Н-дибенз[b,f]азепин (6)
185 г (0,9 моля) 95% иминодибензила (5) растворяют при нагревании в 1500 мл толуола и при 90-95С пропускают через барбатер слабый ток фосгена в течение 3-3,5 ч, затем азотом отдувают избыток фосгена и хлористый водород в течение 2 ч. К раствору прибавляют 14 г активированного угля, кипятят в течение 20 мин, отфильтровывают уголь, промывают 50 мл горячего толуола, фильтрат упаривают до 1/3 объема, охлаждают до 0С, через 4 ч отфильтровывают осадок, промывают 2*40 мл холодного толуола, сушат при 50-60°С и получают 194,4 г кристаллов с т.пл. 121-123°С. Фильтрат упаривают, обрабатывают аналогично активированным углем и выделяют дополнительно 14,2 г кристаллов с т. пл. 119-121°С. Всего получают 208.6 г (90%) (6) (т. пл 121-123°С).
10,11-Дигидро-5-карбамоил-5Н-дибенз[b,f]азепин (9)
Через смесь 38,5 (0,15 моля) (6), 430 мл метанола, 50 мл воды при перемешивании и температуре 57-59°С пропускают ток аммиака в течение 1,5 ч, прибавляют 2 г активированног угля, кипятят в течение 15 мин, фильтруют и фильтрат упаривают досуха. К остаткуприбавляют 400 мл воды, кипятят при перемешивании в течение 1 ч, охлаждают 20°С, отфильтровывают осадок, промывают 10 мл спирта, сушат при 75°С в течение 4 ч и получают 32,7 г вещества с т. пл. 198—205 °С. Осадок перекристаллизовывают из 320 мл метанола с 2 г активированного угля и получают 30,3 г (85.35%) (9) в виде бесцветных кристаллов с т. пл. 205‑206,5°С (206—208°С).
5-Карбамоил-5Н-днбенз[b,f]азепим (4)
Смесь 19,3 г (0,075 моля) (9), 8 мл хлорбензола нагревают до 145°С и при перемешивании при 145-150°С прибавляют по каплям 14,4 г (0,09 моля) брома под слой реакционной массы в течение 1.5 ч с такой скоростью, чтобы холодильник не окрашивался парами брома. Затем перемешивают реакционную массу в течение 2 ч при температуре 150—155°С для завершения дегидробромирования, охлаждают до 90 °С, прибавляют 45 мл метанола, реакционную массу с выпавшим осадком охлаждают до 5°С, отфильтровывают осадок и промывают 2*10 мл метанола. 23,9 г полученной 75% пасты (2) загружают в автоклав емкостью 0,165 л, прибавляют 102 мл метанола, 28,3 мл (0,376 моля) 25% NН4ОН, нагревают при 75-85°С и перемешивании в течение 5 ч, охлаждают до 20°С. выгружают в одногорлую круглодонную колбу, автоклав промывают 50 мл горячего метанола и промывной раствор прибавляют к основной реакционной массе. Смесь кипятят с 4 г активированного угля в течение 30 мин, отфильтровывают уголь, промывают 10 мл кипящего метанола, из фильтрата отгоняют при 64—66°С 150 мл метанола, к остатку прибавляют 20 мл дистиллированной воды, смесь нагревают до кипения и кипятят в течение 1 ч, затем реакционную массу охлаждают до 15°С, осадок отфильтровывают промывают 2*25 мл дистиллированной воды, сушат при 75—80°С в течение 8-10 ч и получают 14,5 г (4) с т. пл. 184—186°С, что составляет 81,87% от теоретического, считая на (6).
Полученные 14,5 г технического карбамазепина (4) загружают в одногорлую круглодонную колбу с обратным холодильником, прибавляют 60 мл спирта, нагревают до растворения осадка, прибавляют 2 г активированного угля, кипятят в течение 30 мин, отфильтровывают уголь, промывают 2*5 мл горячего спирта, фильтрат охлаждают и выдерживают при 0°С в течение 4 ч.
Выпавший осадок отфильтровывают, промывают 2*5 мл холодного спирта, сушат в вакуум-сушильном шкафу при 65-70 °С и получают 10,7 г фармакопейного карбамазепина (4) с т. пл. 190—191,5°С. После упаривания маточного раствора и кристаллизации получают еще дополнительно 1,1 г (4) с т. пл. 189.5—191,5°С. Общий выход (4) 11,8 г, что составляет 66,7% от теоретического, считая на (6), или 60%, считая на исходный (5) [13].
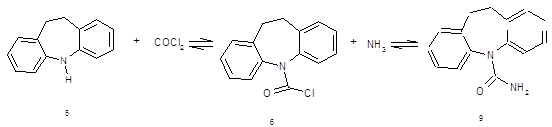
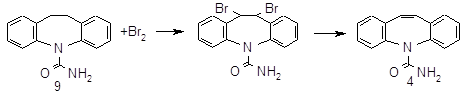
5.3 Баклофен
![]() При обработке 4-ClC6H4COOH (1) и посредством SOCl2 образуется 4‑ClC6H4COCl (2). Восстановлением (2) по Розенмунду получен 4‑ClC6H4CHO (3), конденсация которого с АсCH2COOEt (4) приводит к 4‑ClC6H4CH[CH(Ac)COOEt]2 (5). При щел. гидролизе (5) образуется 4‑ClC6H4CH(CH2 COOН)2 (6), который при кипячении с Ас2О циклизуется в соответствующий ангидрид (7). Реакция (7) с конц. NH4OН приводит к
При обработке 4-ClC6H4COOH (1) и посредством SOCl2 образуется 4‑ClC6H4COCl (2). Восстановлением (2) по Розенмунду получен 4‑ClC6H4CHO (3), конденсация которого с АсCH2COOEt (4) приводит к 4‑ClC6H4CH[CH(Ac)COOEt]2 (5). При щел. гидролизе (5) образуется 4‑ClC6H4CH(CH2 COOН)2 (6), который при кипячении с Ас2О циклизуется в соответствующий ангидрид (7). Реакция (7) с конц. NH4OН приводит к
![]()
4-ClC6H4CHCH2 CONНCOCH2(8)
При обработке (8) посредством Br2 в NaOH образуется 4‑ClC6H4CH(CH2 COOН) CH2NH2 (9).
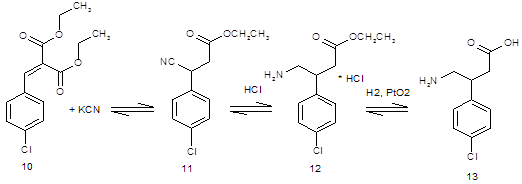
Реакция 4-ClC6H4CH=C(COOEt) 2 (10) с КСN приводит к 4‑ClC6H4CH(CH2COOEt)СN (11), при гидрировании которого образуется 4‑ClC6H4CH(CH2 COOEt)CH2NH2 (12). К-тным гидролизом (12) превращен в 4-ClC6H4CH(CH2COOОН)CH2NH2 (13). При возгонке 4‑Cl‑3‑BrC6H3CH(CH2COOОН)CH2NH2 (14) образуется 4‑(3‑бром‑4хлорфенил)‑2‑пирролидон (15).
При восстановлении (15) посредством Н2 над Pd/C получена смесь 4‑(4‑хлор-3-Н-фенил)-2-пирролидона (16) и 4‑(3,4‑Н2‑фенил)‑2‑пирролидона (17).
Кислотный гидролиз смеси (16) и (17) приводит к смеси:
4-Cl-3-Н-C6H3CH(CH2COOH)CH2NH2(18) и 3,4‑Н2C6H3CH(CH2COOH)CH2NH2 (19).
Раствор 29,1 ммоля (27,3мкюри) (1) в 35 мл SOCl2 кипятят 5ч, р-ритель отгоняют, следы SOCl2 удаляют отгонкой с ГК и получают 5,3 г (2), который используют без очистки на след. стадии.
29,1ммоля (2) гидрируют в слабом токе Н2 в смеси 50 мл ксилола, 0,35г 5% Pd/BaSO4 и 0,021 мл р-ра хинолин-сера до прекращения выделения HCl (70-75С, 28 ч), Кт отфильтровывают, фильтрат упаривают и получают 6,9 г неочищ(3). К р-ру 29,1 ммоль (3)в 4 мл сп.+58,4 ммоль (4) и 1,28 мл пиперидина, выдерж-т 16 ч, прибавляют 50 мл ГК, охлаждают до 0°С и отфильтровывают (5), выход 65%, т. пл. 152-4°С.
Р-р 7,25 г (18,9 ммоля) (5) в 50 мл диоксана прибавляют к смеси 30 мл сп. и 31 г 50% NaOH (100°С, 3ч), выдерживают (100°С, 1ч), охлаждают, орг. р-ритель отгоняют, водн. р-р подкисляют 35 мл конц HCl (0°С), осадок отфильтровывают и получают 3,8 г (6), т. пл. 161-3°С.
Кипятят 3,8 г (6) в 20 мл Ас2О (1ч), р-ритель отгоняют, остаток р-ряют в 5 мл бзл. и высаживают (7) посредством ГК (0°С), получают 3,4 г (7), т. пл. 127-129°С.
К 3,4 г (15,1 ммоля) (7) постепенно прибавляют 15 мл конц. NH4OH (0°С), нагревают (70°С, 0,5 ч), р-ритель отгоняют, остаток нагревают(185°С, 1ч, масляная баня) и получают (8).
15,1 ммоля (8) р-ряют в 16 мл воды, содержащей 0,7 г NaOH (50°С), охлаждают (10-15°С) прибавляют раствор 3,3 г NaOH в 16 мл воды, затем 1,35 мл Br2, выдерживают (20°С, 4 ч), обрабатывают углем, упаривают в вакууме до 20 мл, нейтрализуют до рН 6,5-7 разб. HCl (1:1), осадок отфильтровывают, р-ряют в 15 мл 1н. NaOH, нейтрализуют 1н. HCl до рН 6,5-7, осадок отфильтровывают, сушат в вакууме (25°С/0,01) и получают 1,55г (9) (уд. акт. 4,4 мкюри/ч); из маточного р-ра выделено дополнительное количество (9), общий выход 8,1 мкюри (30%,считая на (1). К р-ру 0,603 г (49,6 кюри) КСN в 1 мл воды прибавляют раствор 2,32 г (9) в 22 мл сп., нагр‑т (70°С, 16 ч, N2), охлаждают (0°С), осадок отфильтровывают, к фильтрату прибавляют 1,6 мл 1н HCl и отгоняют р-ритель. Остаток растворяют в эф., пром-т водой и из орг. слоя выделяют 71,8% (11), т. кип. 140°С/0,05.
Р-р 1,63 г (11) в смеси 11 мл сп. и 0,75 мл 10н. HCl гидрируют над 0,06г PtO2 (20°С, 7 ч), Кт отфильтровывают, фильтрат упаривают, остаток обрабатывают эф. и получают 1,46 г ХГ (12), к-рый кипятят 16 ч с 17 мл 5н. HCl, охлаждают, р-ритель удаляют лиофилизацией, остаток кристаллизуют из смеси сп.-эф. и получают 1,1 г ХГ (13). Выделение и очистку (13) проводят аналогично соединению (9). Получают 19,5 мкюри (13) (39%, считая на КСN).
0,5 г (14) трижды возгоняют (250°С/0,1-0,5) и получают 0,392 г (15), т.пл. 129-32°С/бзл).
Смесь 0,275 г (15), 0,1 г NaОAc, 0,06 г 10% Pd/C и 10мл EtOCH2CH2OH замораживают жидким N2, с-му вакуумируют, заполняют 190 кюри Н2, смесь гидрируют при 20°С (24 мин), избыток Н2 адсорбируют на уране, с-му продувают N2, смесь фильтруют, фильтрат подвергают лиофильной сушке. Остаток раств-т в 10 мл МеОН и вновь лиофилизуют (3 раза). Остаток р-ряют в 10 мл ЭА2 прибавляют 2 мл воды, органический слой отд-т, а водн. экстрагируют 30 мл ЭА, объединенные орг. экстракты пром-т 30 мл воды. Половину полученного раствора лиофилизуют, остаток нагревают в ампуле в 3 мл 6н.HCl(100°С, 2,5 ч), р-ритель отгоняют и хроматографией на СГ в с-ме втор.ВuOH-AcOH-вода (67:10:23) выделяют (18) (3,2 кюри) и (19) (2,4 кюри).
Очистка (18) рехроматогрфией на СГ приводит к 2,03 кюри (18) (уд. радиоакт. 9,38 кюри/ммоль), к-рый хранят в виде р-ра в указанной смеси при пониженной температуре [15].
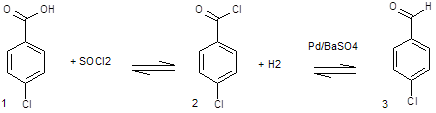
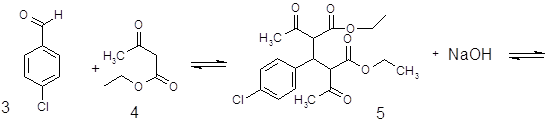
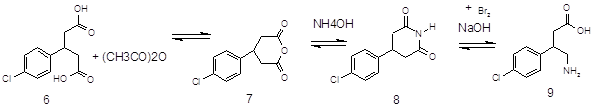
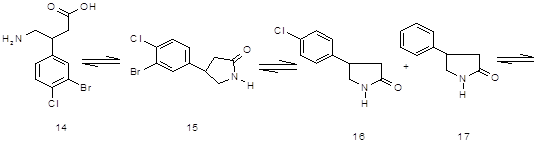
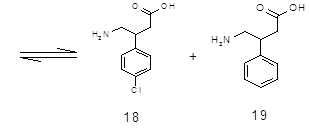
Похожие работы
... заведениях в отсутствии лиц противоположного пола. Это поведение наблюдается у подростков с недостаточно сформированной избирательностью полового влечения. Динамика нарастания делинквентного и девиантного поведения у детей и подростков психиатрического стационара ГУЗ «ОПНБ № 5». 2005 год 2006 год 2007 год Количество поступивших город село город село город село 98 22 122 ...
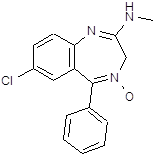
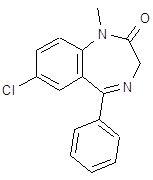
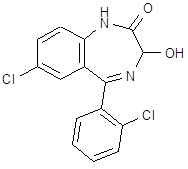
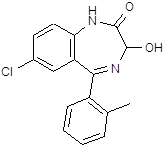
... .Обычно бензодиазепин вводится совместно с совместными анестетиками и наркотиками. Механизм биологической активности [6] Биологическое действие 1,4-бензодиазепинов, как и других лекарственных средств определяется физико-химическим взаимодействием вещества со специфическими молекулярными компонентами в организме, называемыми рецепторами. Продолжительность и интенсивность действия лекарств ...
... в их происхождении. Поэтому широкий арсенал психотропных средств, обладающих воздействием на адреналин-норадреналин, дофамин, серотонин, ГАМК-и нейропептидергические системы, позволяет успешно использовать их в клинической наркологии. Аффективные нарушения при наркоманиях В процессе наркотизации у больных нарастают аффективные расстройства. В течение длительного времени у них преобладает ...
0 комментариев