Навигация
Синдром острого повреждения легких
19981
знак
2
таблицы
0
изображений
Министерство образования Российской Федерации
Пензенский Государственный Университет
Медицинский Институт
Кафедра Хирургии
Зав. кафедрой д.м.н.,
РЕФЕРАТ
на тему:
Синдром острого повреждения легкихВыполнила: студентка V курса
Проверил: к.м.н., доцент
Пенза
2008
План
Введение
1. Патогенез
2. Клиника и диагностика синдрома острого повреждения легких
3. Стадии и периоды
Литература
Введение
В 60-70-х годах разными авторами был описан синдром острой дыхательной недостаточности, возникающий на фоне интенсивной терапии у больных с острой кровопотерей, с тяжелой механической травмой, сепсисом, при применении экстракорпорального кровообращения и т.д. Этот патологический процесс именовался по-разному: “шоковым легким“ (Nickerson M., 1963; Hardaway R.M., 1969; Norlander O., 1975), “септическим легким“ (Collins J.A.,1969), “постперфузионным легочным синдромом“ (Королев Б.А., Шмерельсон М.Б., 1975), “гиповентиляционным диспное“ (Shumer N., 1971) и т.д. На длительное время за ним укрепилось название - “респираторный дистресс-синдром взрослых“, предложенное D.G. Ashbaugh (1967). Впоследствии вместо "взрослых" в название введено определение "острый", поскольку было доказано, что развитие РДС у новорожденных может быть связано с теми же причинами, что и у взрослых.
Классическое клиническое описание ОРДС, описанное D.G. Ashbaugh с коллегами почти 40 лет назад, предусматривает наличие диспноэ, тахипноэ, цианоза, ригидных легких, диффузной альвеолярной инфильтрации. Частота развития ОРДС варьирует от 1,5 до 75 случаев на 100 000 населения в год, что обусловлено различными подходами к его диагностике.
В целом принято считать, что ОРДС характеризуется прогрессирующей гипоксемией и внутрилегочным шунтированием крови, двусторонней инфильтрацией легочных полей на фронтальной рентгенограмме грудной клетки, быстрым снижением податливости легочной ткани, легочной гипертензией при отсутствии признаков левожелудочковой недостаточности. По мере изучения причин развития и патогенеза острого респираторного дистресс-синдрома возникла концепция, согласно которой под ним стали понимать крайнее проявление более широкого процесса, именуемого острым повреждением легких (ОПЛ).
ОПЛ возникает на основе диффузного повреждения эндотелия легочных капилляров под воздействием экзогенных и эндогенных веществ. Развитие тяжелой острой воспалительной реакции и нарушение целостности альвеоло-капиллярной мембраны имеют огромное значение в его патогенезе. Фактически, ОПЛ (ОРДС) является проявлением синдрома общего реактивного воспаления, которое, как сегодня полагают, является основой полиорганной несостоятельности.
1. Патогенез
Патогенез развития ОПЛ и ОРДС во многом еще не изучен, однако ряд важных его звеньев выделить вполне возможно:
- снижение капиллярного кровотока, образование и задержка легочным эндотелием микроагрегатов тромбоцитов и микроэмболов с выходом хемоаттрактантов;
- деструкция задержанных продуктов с образованием биологически агрессивных веществ, повреждающих интерстиций;
- сужение и тромбоз сосудов, часто ведущих к пульмональной гипертензии;
- проникновение богатой протеином жидкости в эктравазальное пространство, отражающее пульмональный отек, не связанный с нарушением функции левого желудочка;
- массивный альвеолярный коллапс, обычно сочетающийся с потерей сурфактанта и снижением растяжимости.
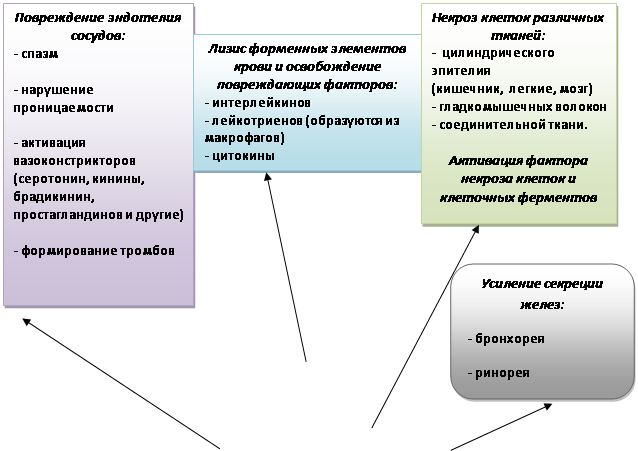
Считается, что в начальной фазе синдрома на фоне замедления кровотока в зоне микроциркуляции легких и образования сначала микроагрегатов тромбоцитов, а затем и микроэмболов происходит адгезия и активация нейтрофилов. Нейтрофилы фиксируются на поврежденном эндотелии легочных капилляров и затем проникают в интерстициальное пространство и просвет альвеол. Из задержанных в легком клеток, главным образом нейтрофилов, освобождаются цитокины, ферменты (эластаза, коллагеназа) и другие вещества, которые повреждают альвеоло-капиллярную мембрану и интерстиций, растворяя эластин, коллаген, фибронектин и т.д. Альвеолярные макрофаги также секретируют цитокины, которые действуют локально, стимулируют хемотаксис и активируют нейтрофилы. Нарушение легочного кровообращения может быть также связано с ролью легких в метаболизме медиаторов воспаления.
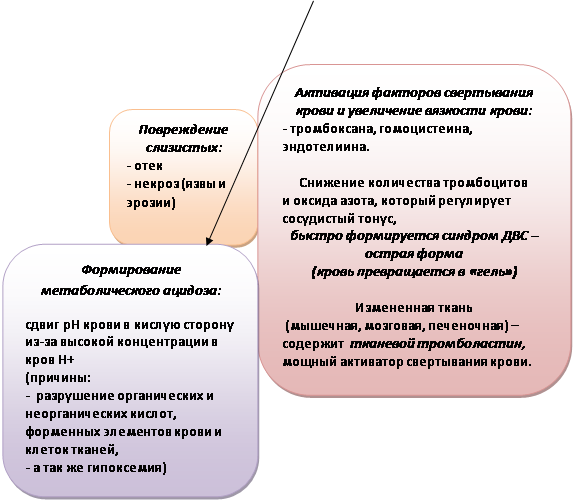
Цитокины (фактор некроза опухоли, интерлейкины) и другие провоспалительные биологически активные вещества являются важнейшими медиаторами воспалительной реакции, инициирующими и усиливающими воспалительный ответ при ОПЛ. Провоспалительные цитокины продуцируются в легких нейтрофилами и альвеолярными макрофагами, эпителиальными клетками и фибробластами. Дальнейшее патологическое воздействие оказывают продукты каскада комплемента, лизосомальные ферменты и биогенные амины, продукты деградации фибриногена и метаболизации арахидоновой кислоты с появлением в циркулирующей крови таких субстанций (эйкосаноиды), как простагландины PgE2 и PgF2, лейкотриены, тромбоксан ТхА2, ФАТ - фактор активации тромбоцитов. Эйкосаноиды не только еще больше увеличивают проницаемость мембраны, но и обладают выраженной бронхо- и вазомоторной активностью; они вызывают бронхиолоспазм, спазм легочных вен и усиливают тромбообразование. Свободные радикалы, выделяющиеся вместе с ферментами, повреждают клеточные мембраны, вызывая пероксидацию липидов, из которых состоят мембраны, а также разрушают гиалуроновую кислоту, связывающую массу соединительной ткани. Проницаемость мембраны возрастает еще более, и этот эффект усиливается при ингаляции 100 % кислорода.
Следствием распространенного повреждения микрососудов становится перемещение значительного количества воды и белка в интерстициальное пространство. Подтверждением этого является нарастание оттока из таких легких тканевой жидкости, богатой протеином: концентрация белка в оттекающей от легких лимфе достигает уровня содержания его в плазме крови. В конечном счете, вследствие нарушения целостности (проницаемости) альвеолокапиллярной мембраны развивается некардиогенный отек легких. Прослеживается прямая связь между степенью функциональной легочной недостаточности в отношении газообмена и внесосудистым объемом воды в паренхиме легких. Альвеолы при этом коллабируются вследствие воспалительного инфильтрата, крови, отечной жидкости, нарушения функции сурфактанта, а также сужения мелких воздухопроводящих путей из-за интерстициального отека и бронхиальной обструкции. В острой фазе синдрома происходят также деструкция альвеолоцитов, сладжирование бронхиальных и альвеолярных клеток, которые при попадании в просвет альвеолы формируют богатые протеином гиалиновые мембраны.
Респироны постепенно заполняются отечной жидкостью; поэтому вначале легочная паренхима представляет мозаику воздушных, коллабированных и отечных конечных легочных единиц. Так, например, при КТ-исследовании легких больного с ОРДС выявляется сочетание коллабированных областей с относительно интактными и вентилируемыми областями. Причем размер и расположение этих зон зависят от используемого ПДКВ, положения тела и других факторов.
Газообмен возможен лишь в непораженных зонах или в участках легких, потенциально включаемых в вентиляцию, например, при использовании ПДКВ. В действительности часто только небольшие участки легкого продолжают вентилироваться. На их долю может приходиться меньше 20 % нормального легочного объема. Перфузия невентилируемых областей является причиной большого шунта справа налево, который может составлять более 60 % сердечного выброса и являться главной причиной артериальной гипоксемии при ОПЛ.
Таким образом, в результате патологических изменений возникает тяжелое несоответствие между вентиляцией и перфузией, ведущее к возникновению шунта справа налево, часто с тяжелой артериальной гипоксемией. Если повреждение капиллярного русла носит системный характер, нарушение доставки кислорода в ткани и нарушение его утилизации в митохондриях приводят к диффузному гипоксическому клеточному повреждению и развитию полиорганной несостоятельности.
Снижение растяжимости легких при ОПЛ происходит вследствие нарушения функции сурфактанта на фоне альвеолярного отека, воспаления, фиброза, влияющих на эластичность легочной ткани. Кроме того, снижение общей растяжимости легких, по крайней мере, отчасти, связано с уменьшением зоны легких, доступных для вентиляции.
Легочная гипертензия обычно развивается у больных с ОПЛ вследствие вазоконстрикции, тромбоза, периваскулярного отека, воспаления, возможного интерстициального фиброза. При выраженной легочной гипертензии может проявиться правожелудочковая недостаточность. Она может быть также следствием сниженной доставки кислорода, неадекватной водной нагрузки, аритмии, истощения, действия миокард-дистрофического фактора на фоне развития системного капиллярного повреждения.
Похожие работы
... развития инфекционно-токсического шока, гиповолемического шока, острой дыхательной недостаточности, полиорганной недостаточности и обострением течения сопутствующих заболеваний. На догоспитальном этапе в оказании экстренной медицинской помощи чаще нуждаются больные с менигококковой инфекцией, острой кишечной инфекцией, тяжелыми и осложненными формами гриппа, дифтерией, малярией, ботулизмом, ...
... дезинтеграция метаболических процессов, участвующих в формировании долговременной адаптации, усугубляется феноменом взаимного отягощения. Наиболее клинически манифестированным проявлением третьего периода травматической болезни – периода максимального развития осложнений - является синдром полиорганной недостаточности. Причинами ее являются длительная гипотензия, массивные гемотрансфузии, ...


... попадании в организм пищевого аллергена. В каждой форме доминируют свои признаки, которые заложены в названии. После анафилактического шока развиваются осложнения: · Острый миокардит · Гломерулонефрит · ДВС- синдром · Инфаркт миокарда · Гемолитическая анемия · Менингоэнцефалит · Арахноидит · Полиартрит · Параличи и парезы из-за тромбозов сосудов мозга · ОПН Причины смерти ...
... . Предельно допустимым является максимальное пиковое давление паузы вдоха около 35 см Н2О. 4. Обоснованное использование фармакологическое угнетения сознания и мышечного тонуса пациента с обязательным мониторингом состояния больного. При этом руководствуются показателями доставки кислорода, степенью гипоксемии, переносимостью данного режима ИВЛ. При терапии ОРДС в последние годы все шире ...
0 комментариев