Навигация
Спадкові хвороби. Спадкові хвороби обміну речовин
25852
знака
0
таблиц
2
изображения
МІНІСТЕРСТВО ОХОРОНИ ЗДОРОВ'Я УКРАЇНИ
ЛУБЕНСЬКЕ МЕДИЧНЕ УЧИЛИЩЕ
РЕФЕРАТ
З ПЕДІАТРІЇ
НА ТЕМУ: Спадкові хвороби. Спадкові хвороби обміну речовин
1 Спадкові хвороби
Медико-генетичне консультування
Генетична консультація надає допомогу у вирішенні проблем, пов'язаних з ризиком генетичних захворювань. До генетичної консультації найчастіше звертаються подружжя, в яких перша дитина народилася з вродженим дефектом чи з іншою патологією.
В ідеальних умовах подружжю слід звернутися до генетичної консультації та обговорити з лікарем ризик народження дитини з вродженою аномалією ще до зачаття чи в перші 3 міс вагітності.
Існують категорії сімей, яким необхідне генетичне консультування, навіть якщо в роду нема випадків спадкової патології. Це сім'ї, в яких один або обидва партнери мають шкідливі звички, професійні шкідливості; якщо жінка старша 35-річного віку, а також коли в анамнезі були спонтанні викидні, мертвонародження.
Слід вважати обов'язковим для кожної родини з встановленими випадками спадкової хвороби звертатися до генетичної консультації для обговорення питання про планування сім'ї.
Основними задачами генетичного консультування є такі:
1)уточнення генетичного діагнозу та визначення індивідуальної величини генетичного ризику для конкретної родини;
2)вирішення питання про планування вагітності та вибір методу пренатальної діагностики (якщо метод інформативний у даному випадку).
Першим етапом генетичного консультування є складання генетичного дерева та аналіз походження. Необхідно зібрати повну інформацію про стан здоров'я всіх членів родини, з урахуванням кровної родинності, національності, наявності вроджених вад чи викиднів у членів родини (не менше ніж у 4 поколінь).
При виявленні захворювання необхідна точна діагностика, щоб виявити його природу, провести диференціальну діагностику станів, що мають подібний клінічний вигляд. Якщо при дослідженні родоводу з'ясовані не всі випадки захворювань, то не завжди можна виявити тип успадкування (наприклад, аутосомно-рецесивний чи рецесивний X-зчеплений тип). Значно допомагає визначити тип спадкової хвороби виявлення гетерозиготного носійства мутантних генів родичами хворого (за допомогою лабораторних, найчастіше біохімічних методів).
При встановленні точного діагнозу стає можливим розрахувати ймовірність народження дитини зі спадковою патологією. Якщо сімейна пара вже має дитину з установленим спадковим захворюванням, ризик виникнення повторного захворювання (зворотний ризик) складається вже з відомого для даного захворювання ризику і загальнопо-пуляційного ризику (3 % для всіх інших вроджених дефектів). Необхідно враховувати, що для будь-якої вагітності існує ймовірність виникнення вродженої вади, яка дорівнює 3 %.
Генетичний ризик прийнято кваліфікувати так: до 5 % —низький, до 10 % — незначно підвищений, до 20 % — середній, понад 20 % — високий.
У разі підвищеного ризику хромосомних та багатьох моногенно успадкованих захворювань виправдане використання інвазивних методів перинатальної діагностики.
Преімплантаційна пренатальна діагностика
В Україні преімплантаційна пренатальна діагностика перебуває на стадії експериментального введення. Під час цього дослідження вивчають запліднену яйцеклітину на стадії 8 бластомер (12 год після запліднення). ДНК тиражують у мільйонах копій за допомогою молекулярно-генетичного методу полімеразної ланцюгової реакції.
Перинатальна діагностика
Перинатальна діагностика дозволяє встановити наявність вродженої вади чи генетичного захворювання плода на ранніх етапах його розвитку.
Своєчасне виявлення вродженої вади дозволяє прийняти рішення про припинення вагітності чи морально підготуватися до народження хворої дитини. Крім того, рання перинатальна діагностика дозволяє адекватно контролювати перебіг вагітності, пологів та неонаталь-ного періоду.
До методів перинатальної діагностики належать такі:
1)неінвазивні:
а) визначення рівня альфа-фетопротеїну;
б) ультразвукове дослідження;
2) інвазивні методи:
а) амніоцентез;
б) біопсія ворсин хоріона;
в) фетоскопія.
Визначення рівня альфа-фетопротеїну сироватки крові матері проводять на 16—18-му тижні вагітності.
Підвищення рівня альфа-фетопротеїну може свідчити про наявність вад розвитку центральної нервової системи плода, дефектів передньої черевної стінки (наприклад омфалоцеле), відшарування плаценти. Діагностично інформативним прийнято вважати підвищення рівня альфа-фетопротеїну в 2,5 разу і більше від середнього значення для конкретного терміну вагітності, особливо при поєднанні а Підвищенням вмісту іншого сироваткового маркера — хоріонального Гонадотропіну.
Знижений рівень альфа-фетопротеїну може спостерігатися при хворобі Дауна та інших трисоміях.
Ультразвукове дослідження — безпечний метод перинатальної діагностики, який може бути використаний за підозри на наявність структурних аномалій. Вважається, що воно не справляє ризику для матері 1 та плода. При масовому ультразвуковому скринінгу вагітних звичайно Визначають розміри та масу тіла плода, їх кількість і життєздатність. Також проводять оцінку стану плаценти, пуповини, навколоплідних Вод. Під час ультразвукового дослідження може бути виявлена більшість структурних аномалій основних органів і систем у термін гестації до 16—18 тиж, а при деякій патології і раніше. У цих випадках показано ретельне ультразвукове дослідження в комплексі з амніоцентезом [та біопсією ворсин хоріона.
Перед тим як вирішити питання про необхідність інвазивного методу діагностики, необхідно врахувати такі умови:
а) чи достатньо тяжким є захворювання, щоб проводити втручання;
б) чи існують чіткі тести для пренатальної діагностики захворю
вання;
в) чи дійсно генетичний ризик настільки значний, щоб виправдати
ризик проведення інвазивного методу діагностики.
Амніоцентез. Звичайно амніоцентез проводять на 16-му тижні вагітності. Припустимі і більш ранні терміни (13—15-й тиждень) з ретельним дотриманням заходів безпеки.
За допомогою спеціальної голки, якою проникають в амніотичний Простір, отримують близько ЗО мл амніотичної рідини. Дослідження амніотичної рідини включає:
—хромосомний аналіз;
—визначення рівня альфа-фетопротеїну;
—дослідження клітинного складу;
—дослідження ДНК;
—визначення біохімічних маркерів;
—визначення статі плода (якщо мати — носій захворювання, зчепленого з Х-хромосомою, то переривання вагітності при встановленні Чоловічої статі плода дозволить запобігти народженню хворої дитини).
При амніоцентезі ризик ускладнень становить 0,25—0,5 % . Зокрема це:
—самовільні потуги, які призводять до аборту;
—підтікання навколоплідної рідини, що призводить до маловоддя або спонтанних пологів;
—травмування плода голкою в момент пункції (дуже рідко);
—— інфікування (дуже рідко, при порушенні правил асептики).
Показаннями до проведвагітні віком понад 35 років;
—вагітні з підвищеним рівнем альфа-фетопротеїну в крові;
—вагітні зі зниженим рівнем альфа-фетопротеїну в крові;
—жінки, яки мають дитину з хворобою Дауна (зворотний ризик 1—2%);
—сім'ї, в яких один з батьків — носій симетричної реципрокноі транслокації хромосоми, мають високий ризик народження дитини н асиметричною транслокацією;
—сім'ї, у родоводі яких виявлені захворювання з аутосомним X зчепленим типом успадкування.
Біопсія ворсин хоріона. Пункція ворсин хоріона — відносно новий метод перинатальної діагностики, який дозволяє проводити дослід ження на 8— 11 -му тижні вагітності.
Плаценту пунктують спеціальною голкою через передню черевну стінку (під контролем ультразвукового дослідження). Отримані при аспірації клітини ворсин хоріона підлягають різнобічному досліджен ню. Зокрема проводять:
—хромосомний аналіз;
—дослідження рівня ферментів (якщо в плода є ймовірність роп витку захворювання, пов'язаного з недостатністю конкретного фер менту);
—дослідження ДНК (якщо в плода є ризик виникнення моноген ного захворювання з відомим генним дефектом);
—визначення статі плода (при загрозі успадкування захворювап ня, зчепленого зі статтю).
При біопсії ворсин хоріона ризик ускладнень високий — 1—5 %. Порівняно з амніоцентезом загроза переривання вагітності або внутрі-шньоутробного інфікування (хоріонамніоніт) значно вищі.
Перевагою методу є його інформативність на ранніх етапах розвит ку плода (до 12-го тижня вагітності).
Недоліками методу є те, що при біопсії ворсин хоріона не можнй оцінити рівень альфа-фетопротеїну, тому що процедура проводиться на ранніх термінах формування плода і дослідженню доступні лиш* клітини; у деяких випадках після біопсії ворсин хоріона виникає іір обхідність додаткового проведення амніоцентезу (наприклад, при мо-заїцизмі).
Фетоскопія. Під контролем ультразвукового дослідження за допомогою тонкого оптично-волоконного лапароскопа через передню черевну стінку проникають у порожнину матки — з'являється можливість спостереження і дослідження плода. Можлива біопсія шкіри плоди для діагностики спадкових захворювань шкіри (наприклад іхтіозу),
Із судин пуповини можна забрати кров для діагностики гемоглобі нопатій чи інших хвороб крові. Кров забирають під контролем ультря звукового дослідження (кордоцентез). Метод використовують для ення амніоцентезу є такі групи ризику:
хромосомного дослідження та біохімічного аналізу крові плода (наприклад, за підозри на гемофілію).
Хвороба (синдром) Марфана
Хвороба Марфана діагностується з частотою 1 на 20 000 новонароджених. Тип успадкування аутосомно-домінантний. Виникнення синдрому обумовлене мутацією в гені, відповідальному за синтез фібри-ліну, одного із структурних білків основної речовини сполучної тканини, що призводить до різних аномалій розвитку. При синдромі Марфана виявлено порушення обміну кислих полісахаридів (глікоза-МІногліканів) типу хондроїтинсірчаної та гіалуронової кислот як у Волокнах, так і в основній речовині сполучної тканини і виведення їх Із сечею. Також порушується обмін оксипроліну — важливого компоненту колагену. Наведені біохімічні дані можуть слугувати основою для підтвердження синдрому Марфана. Як правило, у 70—80 % хворих один з батьків страждає на даний синдром. У той самий час частота нових мутацій досить висока.
У типових випадках захворювання в пацієнта наявна тріада клінічних симптомів, які дають можливість діагностувати даний синдром: ураження скелета, серцево-судинної системи та очей. У хворих відзначають астенічну будову тіла, довгі й тонкі кінцівки, арахнодактилію кистей і стоп, лійкоподібну деформацію грудної клітки, кіфози, кіфо-околіози, вузький лицевий скелет, високий зріст, слабкість зв'язкового апарату.
Ураження очей можуть бути різноманітними — катаракта, вторинна глаукома, гетерохромія райдужки тощо, але найтиповішими є вивих і підвивих кришталика й обумовлена цим міопія високого ступеня або гіперметропія.
Серцево-судинні прояви захворювання в дітей часто виявляють у вигляді порушень провідної системи серця, пролапсу мітрального клапана, метаболічних змін у міокарді, у дорослих — аневризми аорти, які можуть бути причиною раптової смерті хворих. Як правило, у Хворих має місце дилатація устя аорти, що призводить до аортальної Недостатності, а внаслідок ураження середньої оболонки аорти може відбуватися розшарування стінки з утворенням аневризми. Окрім Того, у дорослих відзначають розвиток емфіземи легень, легеневих бул в можливим їх подальшим розривом — це все наслідок дегенеративних змін легеневої тканини.
Перебіг хвороби в більшості хворих дітей сприятливий. Однак з віком прогресують кількісні та якісні зміни в білковому обміні, особливо в обміні глікозаміногліканів. Декомпенсація патологічного процесу настає після 40-річного віку, коли розвиваються явища серцевої недостатності, розриви судинних аневризм.
Своєчасна діагностика хвороби Марфана дозволяє проводити об ґрунтоване медико-генетичне консультування і прогнозування. Лікування лише симптоматичне.
Синдром Клайнфельтера
Захворювання входить до числа хромосомних хвороб, а саме до аномалій статевих хромосом. Каріотип при даному синдромі — 47, XXY (можливі випадки з наявністю ще більшого числа Х-хромосом). Захворювання зустрічається з частотою 1 на 500 новонароджених хлопчиків.
Наявність зайвої (або зайвих) Х-хромосом, що спричинене нероз ходженням їх при гаметогенезі, сприяє гіалінозу сімейних канальців з подальшою азоспермією при нормальному розвитку чоловічих гені талій і вторинних статевих ознак. У хворих при різко зниженому спер матогенезі підвищується активність гонадотропних гормонів, що сприяє збільшенню кількості клітин Лейдіга, які, крім андрогенів, сприяють утворенню значної кількості жіночих статевих гормонів, що зумовлює генікомастію хлопчиків.
Хворі з синдромом Клайнфельтера мають високий зріст, довгі кінцівки, слабко розвинені м'язи. Зовнішні статеві органи в них розвинені за чоловічим типом, але яєчка маленькі й дуже щільні. Постійною ознакою в подальшому є неплідність, лише в деяких випадках може визначатися сперматогенез. У період статевого розвитку частим симптомом є двобічна гінекомастія, жіночий тип оволосіння і відкладення підшкірної жирової клітковини за жіночим типом.
Лікування остаточно не розроблене. Хворим з нормальним розвитком інтелекту і правильною поведінкою в період статевого розвитку проводять лікування андрогенами. Гінекомастія часто лікується хірургічним шляхом.
Синдром Шерешевського—Тернера
Захворювання пов'язане з аномалією кількості статевих хромосомі Зустрічається з частотою 1 : 2500 осіб жіночої статі. Каріотип — 45, Х0. В основі захворювання має місце патологічний набір хромосом і дефіцитом однієї Х-хромосоми. У низці випадків спостерігаються лише морфологічні зміни в Х-хромосомах при нормальній їх кількості,
Захворювання характеризується вираженим статевим інфантилів» мом, низькорослістю, cubitus valgus, утворенням типових складок Ні бічних поверхнях шиї. Низькорослість хворих помірна, у них широка грудна клітка і велика відстань між сосками. Ріст дітей значно упо» вільнений, кінцевий зріст рідко перевищує 135 см, але низько1 рослість менша, ніж при гіпофізарному нанізмі. У хворих можливі такі аномалії розвитку, як деформації вушних раковин і їх низьке роа» ташування, зменшена нижня щелепа, епікантус (третя повіки), полідактилія, синдактилія та ін. Досить часто при цьому синдромі!
вади розвитку внутрішніх органів: коарктація аорти, відкрита артеріальна протока, декстракардія, аномалії нирок. Розумовий розвиток переважно нормальний.
Найбільш постійним симптомом є дисгенезія гонад. У пацієнток немає яєчників, вони замінені рудиментарними тяжами зі сполучної тканини. Порушення розвитку статевої системи проявляються інфантилізмом. Такі хворі, як правило, неплідні, у них слабко розвинені вторинні статеві ознаки, немає менархе. Особливістю проявів хвороби у дітей грудного віку є наявність лімфатичного набряку стоп, гомілок і кистей, а також крилоподібних складок на шиї.
Диференціальну діагностику в нетипових випадках хвороби необхідно проводити з ювенільною мікседемою, гіпофізарним нанізмом.
Лікування симптоматичне. Призначають анаболічні гормони для стимуляції росту, а при досягненні віку 13—14 років — естрогени. При значно виражених шийних складках показана пластична операція.
Хвороба Дауна
Серед синдромів трисомій аутосомна трисомія 21+ трапляється найчастіше. Діагностується 1 випадок на 700 живонароджених дітей. Патологія зустрічається однаково часто у хлопчиків і дівчаток. У 95 % хворих виявляють 47 хромосом, при цьому 3 з них — у 21-й парі. Таке порушення каріотипу відбувається у випадку, коли в матері при дозріванні статевої клітини під впливом несприятливих факторів відбувається нерозходження 21-ї пари хромосом і утворюється яйцеклітина, яка має 24 хромосоми. При заплідненні такої яйцеклітини нормальним сперматозоїдом, який має 23 хромосоми, розвивається плід з 47 хромосомами. У частини хворих (3—5 %) каріотип має 46 хромосом, але мають місце порушення структури 21-ї хромосоми (транслокація).
Клінічні ознаки хвороби Дауна в типових випадках дозволяють діагностувати захворювання вже на ранніх етапах життя дитини (плода). Типовими ознаками є малий зріст, маленька округла голова зі скошеною потилицею, амімічне обличчя, косий розріз очей, епікантус, плоске і широке перенісся, маленькі деформовані вушні раковини, м'язова гіпотонія, поперечна шкірно-підшкірна складка на потилиці. Кисті рук плоскі, пальці короткі, є великий проміжок між І і II пальцями. Типовим для захворювання є долонний малюнок складок шкіри. Відомо, що в нормі серед численних м'яких складочок виділяють три найбільш великі, одна з яких у вигляді дуги проходить навколо підвищення основи великого пальця, а дві інші перетинають долоню у поперечному напрямку. При хворобі Дауна обидві поперечні лінії зливаються в одну, яка проходить через усю долоню. Хворі відстають у фізичному й особливо нервово-психічному розвитку. Розумове відставання характеризується імбецильністю різних ступенів. У 40—50 % хворих виявляють вроджені вади серця і судин та інші аномалії.
Лікування симптоматичне.
Похожие работы
... і передається також зміни активності ферментів, що прискорюють жировий обмін та згоряння жиру в організмі, а також особливості нейроендокринної системи та певна спрямованість білкового та вуглеводного обміну речовин. Значну роль генетичного фактору у виникненні ожиріння підтверджують результати дослідження, проведеного у Лавальському університеті Квебеку. Там досліджувалися молоді дорослі чолов ...
... які передаються нащадкам разом з нормальними генами. Якщо припустити, що кожна хвороба зумовлена порушенням функції кількох генів, то можна вважати, що у кожної людини близько 1 % генів зачеплена мутацією. Усі спадкові захворювання можна поділити на три групи: 1. Моногенні, або молекулярні. 2. Хромосомні. 3. Полігенні, або полі факторіальні. Спадкові захворювання виникають внаслідок змін ...
... уникнути перегрівання на сонці, треба купатись ранком (з 7 до 10 годин) або в передвечір'я (з 17 до 19 годин). Морські хвилі, масажуючи тіло, тренують серце і судини. Купатися в морі хворим на гіпертонічну хворобу дозволяється через 3—5 днів адаптації до курортних умов. Ослаблені і схильні до простуди люди повинні попередньо пройти підготовку за допомогою різних загартовуючих процедур (прийняття ...
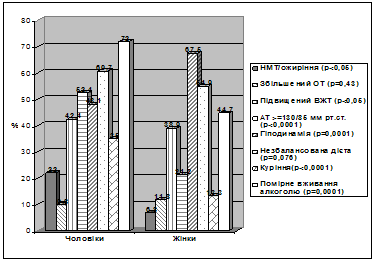
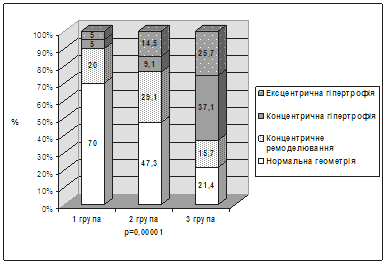
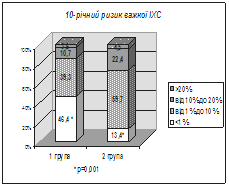
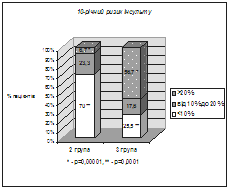
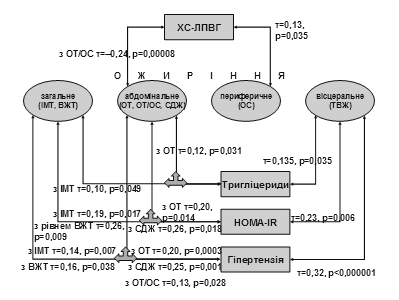
... І., Томашевська О. Я. – К. : Гідромакс, 2006. – 138 с. (Здобувач опрацювала принципи та апробувала схему гемостазіологічного дослідження, написала розділи посібника). АНОТАЦІЯ Томашевська О.Я. Зв'язок між метаболічним синдромом та виникненням цукрового діабету й серцево-судинних хвороб: клініко-лабораторні та функціональні предиктори. – Рукопис. Дисертація на здобуття наукового ступеня
0 комментариев