Навигация
Спадкові хвороби обміну речовин
25852
знака
0
таблиц
2
изображения
2 Спадкові хвороби обміну речовин

Основні клінічні ознаки вроджених порушень метаболізму
Рецидивуючі напади блювання, ацидоз (після початку годування грудним молоком або молочними сумішами) можуть вказувати на по> рушення метаболізму амінокислот або вуглеводів.
![]() Незвичайний запах сечі та поту може зустрічатися при деяких станах, наприклад, при хворобі кленового сиропу; мишачий запах — при фенілкетонурії.
Незвичайний запах сечі та поту може зустрічатися при деяких станах, наприклад, при хворобі кленового сиропу; мишачий запах — при фенілкетонурії.
Гепатоспленомегалія можлива при хворобах накопичення, коли метаболіти відкладаються в клітинах печінки та в селезінці.
Неврологічні розлади носять генералізований характер, можуть бути розлади руху, судоми.
Затримка розвитку інтелекту наявна при фенілкетонурії, мукопо-лісахаридозах.
Ознаками вроджених порушень метаболізму можуть бути також міопатія, дилатаційна кардіоміопатія, затримка росту, виражений ацидоз при аміноацидуріях, гіперамоніємія (пов'язана з порушенням обміну сечовини й органічних кислот), а також наявність в сімейному анамнезі випадків смерті в ранньому дитинстві.
Лікування гострих захворювань у випадках, коли не можна виключити спадкові хвороби обміну речовин
Гострі метаболічні розлади можуть швидко прогресувати й загрожувати життю. Лікування починають ще до встановлення точного діагнозу.
1.Припиняють ентеральне годування, доки не буде встановлений точний діагноз і відкоригована дієта.
2.Підтримують дихання і кровообіг, усувають метаболічний ацидоз і гіперамоніємію. При тяжкій гіперамоніємії показаний діаліз. З лікарських засобів застосовують бензойну кислоту, фенілацетат (на стадії клінічних випробувань).
3.Збирають матеріал для досліджень (кров, сечу, випорожнення, спинномозкову рідину та ін.).
4.Для зниження катаболізму внутрішньовенно вводять 10 % розчин глюкози. Жирові емульсії необхідно застосовувати з обережністю. Інфузійні розчини, що містять лактат, виключають через небезпеку розвитку лактацидозу при мітохондріальних захворюваннях.
5.Виведенню органічних кислот сприяють вітаміни, жирів — карні-тин, а також діаліз, гемодіаліз.
6.При розвитку сепсису (при галактоземії) проводять його лікування.
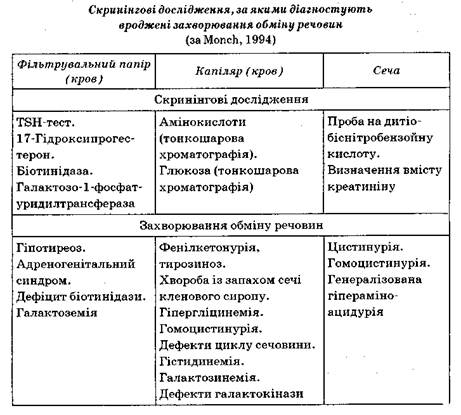
Діагностичні можливості скринінгу вроджених захворювань обміну речовин
Скринінг вроджених захворювань обміну речовин повинен проводитися всім новонародженим дітям. Для дослідження беруть кров (у капіляр), сечу, а також 6 крапель крові на спеціальний фільтрувальний папір. Матеріал для дослідження беруть на 3-тю — 6-ту добу після народження. Для амінокислотного аналізу і дослідження вмісту цук
ру в сучасних умовах рекомендована тонкошарова хроматографія, тест
Guthrie (мікробіологічний тест гальмування) менш інформативний.
При скринінгових дослідженнях проводять діагностику таких за
хворювань.
Коротка характеристика спадкових захворювань обміну речовин, що найчастіше зустрічаються
Фенілкетонурія — це спадкове аутосомно-рецесивне генетичне захворювання, пов'язане з порушенням амінокислотного метаболізму, що призводить до ураження головним чином центральної нервової системи, Частота фенілкетонурії серед новонароджених становить від 1: 5000 до 1 : 10 000. Хворіють хлопчики та дівчатка, але дівчатка дещо частішої Виникає внаслідок мутації гена печінкового фермента фенілаланінгід-роксилази. Цей фермент каталізує окислення фенілаланіну в тирозин. Патологічні зміни при фенілкетонурії пов'язують з накопиченням фені* лаланіну і повторними порушеннями обміну інших амінокислот.
![]()
![]()
![]() Клінічні симптоми з'являються ближче до другого півріччя життя. Фізичний розвиток знижений, відзначається зменшення розмірів черепа, пізнє прорізування зубів, аномалії скелета.
Клінічні симптоми з'являються ближче до другого півріччя життя. Фізичний розвиток знижений, відзначається зменшення розмірів черепа, пізнє прорізування зубів, аномалії скелета.
Моторний розвиток: діти пізно починають сидіти, ходити, мають своєрідну ходу, сидять підібравши ноги в «положенні кравця» у зв'язку з гіпертонією м'язів,
Психічний розвиток: при відсутності лікування в 60 % дітей розвивається ідіотія, приблизно у 10 % — слабкий ступінь олігофренії. Шкіра позбавлена пігментації, волосся світле, очі блакитні, підвищена чутливість до сонячних променів, діти схильні до частих травм. Характерні екзема, дерматит, папульозний висип.
Нервова система: епілептичні напади, атаксія, гіперкінези, тремор, м'язові судоми, тремтіння, сухожилкові рефлекси підвищені, позитивний симптом Бабінського, дермографізм посилений, виражена пітливість, акроціаноз. Артеріальний тиск знижений. Відзначаються закрепи. Сеча має мишачий запах.
Діагноз встановлюють на основі клініки та підтвердження спеціальними пробами наявності фенілаланіну в сечі та крові (тест Феллінга, проба Гатрі, тонкошарова хроматографія).
Лікування: призначення дієти (заміщення природних натуральних білків препаратами зі зниженим вмістом фенілаланіну: білкові гідролізати «Берлофен», «Нофемікс», «Нофелан»).
Галактоземія — захворювання, пов'язане зі спадковим дефектом обміну вуглеводів.
Розвивається у зв'язку з неможливістю використання організмом галактози й проявляється у вигляді тяжкого ураження печінки, нервової системи, очей та інших органів. За частотою посідає 2-ге місце після фенілкетонурії. Тип успадкування аутосомно-рецесивний. В основі захворювання лежить відсутність або різке зниження активності ферменту галактозо-1-фосфат-уридилтрансферази, що є необхідним на другому етапі Перетворення галактози на глюкозу. Внаслідок цього нагромаджуються продукти обміну галактози — галактозо-1-фосфат. Це призводить до токсичної дії на нервову систему, можливості набряку мозку через високу концентрацію галактози в спинномозковій рідині та шлуночках мозку. Нерідко відзначаються гіпоглікемія, ураження печінки, що супроводжуються жовтяницею, підвищенням рівня прямого білірубіну в крові, гепатомегалією, можлива гемолітична анемія. Шлунково-кишкові розлади характеризуються блюванням, зригуванням (з моменту введення молока), що може призводити до гіпотрофії. Лікування полягає у призначенні безмолочної дієти (мигдалеве Молоко, казеїнові гідролізати з видаленою лактозою). З раціону виключають молоко мінімум на 3 роки. Прогноз може бути добрим. Хворі, що не одержують адекватної терапії, частіше вмирають у грудному віці від кахексії і колібацилярного сепсису.
Глікогенози, тобто хвороби накопичення глікогену, — група вроджених захворювань, що розвиваються при недостатності ряду ферментів, унаслідок чого відбувається накопичення глікогену. Залежно від характеру ензимного дефекту виділяють 12 типів глікогенозів. Тип успадкування — аутосомно-рецесивний; частота — 1: 68 000.
I тип — хвороба фон Гірке, розвивається внаслідок дефіциту фер
менту глюкозо-6-фосфатази, що забезпечує нормальний розпад гліко
гену до глюкози. Немобілізований глікоген нагромаджується в значній
кількості в печінці й дещо менше в нирках, що зумовлює глікогенну
гепатонефромегалію. Маніфестує в грудному віці гепато- і нефроме-
галією, гіпоглікемією, ацидозом. Часто визначається гіперкетонемія.
При біопсії печінки визначають високий рівень глікогену, знижену
активність глюкозо-6-фосфатази.
Терапія спрямована на запобігання гіпоглікемії. Призначається дієта, багата на вуглеводи (наприклад кукурудзяний крохмаль).
II тип — глікогенна кардіомегалія (генералізований глікогеноз,
хвороба Помпе). Характерне поширене відкладення глікогену в
печінці, нирках, міокарді, нервовій системі, скелетних м'язах та ін.
Ефективного лікування досі не знайдено.
Мукополісахаридози — група захворювань, що характеризуються дефіцитом лізосомальних ферментів метаболізму мукополісахаридів. Усі (крім синдрому Хантера) мають аутосомно-рецесивний тип успад кування. Частота — 1 : 25 000. Симптоми захворювання виникають унаслідок внутрішньоклітинного накопичення мукополісахаридів і виявляються не відразу після народження. Хвороба уражує всі органи й тканини. Але більшою мірою уражуються печінка і селезінка (ге-патоспленомегалія), розвиток скелета (дисплазія скелета, карликовість, контрактури суглобів), головний мозок (мегаенцефалія, затримка інтелектуального розвитку), серце (аортальна і мітральна недостатність) і дихальна система (стеноз трахеї). Мукополісахаридози звичайно поєднуються з прогресуючими дефектами неврологічних функцій і зниженням інтелекту. При деяких формах мукополісаха ридозів мозок залишається інтактним і інтелект збереженим.
Хвороба Гоше належить до групи спадкових порушень метаболізму ліпідів. Характеризується зниженим рівнем ферменту глюкоцереб розидази. Дефіцит глюкоцереброзидази призводить до акумуляції ліпіду глюкоцереброзиду в лізосомах клітин моноцитарно-макрофа-гальної системи. Навантажені ліпідами клітини з ексцентрично роа-ташованою цитоплазмою відомі як клітини Гоше і становлять первинний субстрат хвороби. Надлишок клітин Гоше призводить до заміщеи* ня нормальних здорових клітин кісткового мозку, гепатомегаліїї дисфункції органів, погіршення стану кісткової тканини. Симптоми" ми захворювання є анемія, тромбоцитопенія, затримка росту.
Методи діагностики — дослідження кісткового мозку (виявлення клітин Гоше), дослідження вмісту глюкоцереброзидази в лейкоцитах, аналіз на основі дослідження ДНК.
Лікування: замісна терапія ферментом «Цередаза» або «Церезим», які є модифікованими формами глюкоцереброзидази.
Використана література
1. Педіатрія: Навч. посібник / О.В. Тяжка, О.П. Вінницька, Т.І. Лутай та ін.; За ред. проф. О.В. Тяжкої. — К.: Медицина, 2005. — 552 с.
Похожие работы
... і передається також зміни активності ферментів, що прискорюють жировий обмін та згоряння жиру в організмі, а також особливості нейроендокринної системи та певна спрямованість білкового та вуглеводного обміну речовин. Значну роль генетичного фактору у виникненні ожиріння підтверджують результати дослідження, проведеного у Лавальському університеті Квебеку. Там досліджувалися молоді дорослі чолов ...
... які передаються нащадкам разом з нормальними генами. Якщо припустити, що кожна хвороба зумовлена порушенням функції кількох генів, то можна вважати, що у кожної людини близько 1 % генів зачеплена мутацією. Усі спадкові захворювання можна поділити на три групи: 1. Моногенні, або молекулярні. 2. Хромосомні. 3. Полігенні, або полі факторіальні. Спадкові захворювання виникають внаслідок змін ...
... уникнути перегрівання на сонці, треба купатись ранком (з 7 до 10 годин) або в передвечір'я (з 17 до 19 годин). Морські хвилі, масажуючи тіло, тренують серце і судини. Купатися в морі хворим на гіпертонічну хворобу дозволяється через 3—5 днів адаптації до курортних умов. Ослаблені і схильні до простуди люди повинні попередньо пройти підготовку за допомогою різних загартовуючих процедур (прийняття ...
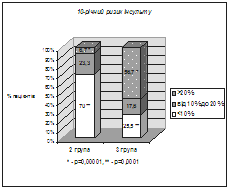
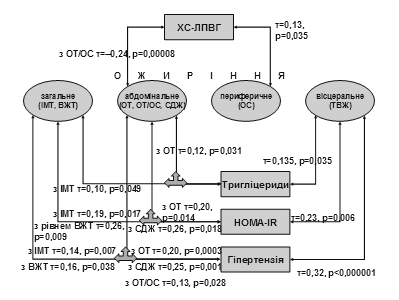
... І., Томашевська О. Я. – К. : Гідромакс, 2006. – 138 с. (Здобувач опрацювала принципи та апробувала схему гемостазіологічного дослідження, написала розділи посібника). АНОТАЦІЯ Томашевська О.Я. Зв'язок між метаболічним синдромом та виникненням цукрового діабету й серцево-судинних хвороб: клініко-лабораторні та функціональні предиктори. – Рукопис. Дисертація на здобуття наукового ступеня
0 комментариев