Навигация
Точка S – проверка точности репликации
33158
знаков
3
таблицы
2
изображения
2. Точка S – проверка точности репликации.
3. Точка G2/M‑перехода – проверка завершения репликации.
4. Переход от метафазы к анафазе митоза.
Перед началом репликации Sc ORC‑комплекс (origin recognition complex) садится на ori – точку начала репликации. Cdc6 представлен во всем клеточном цикле, но его концентрация возрастает вначале G1, где он связывается c ОRC комплексом, к которому затем присоединяются Mcm белки с образованием pre-replicative complex (pre-RC). После сборки pre-RC клетка готова к репликации.
Для инициации репликации S-Cdk соединяется с протеинкиназой (?), которая фосфорилирует pre-RC. При этом Cdc6 диссоциирует от ОRC после начала репликации и фосфорилируется, после чего убиквитинируется SCF и деградирует. Изменения в pre-RC препятствуют повторному запуску репликации. S-Cdk так же фосфорилирует некоторые Mcm белковые комплексы, что запускает их экспорт из ядра. Последующая дефосфориляция белков вновь запустит процесс образования pre-RC.
Циклины – активаторы Cdk. Циклины, так же как и Cdk вовлечены в различные, помимо контроля клеточного цикла, процессы. Циклины разделяются на 4 класса в зависимости от времени действия в клеточном цикле: G1/S, S, M и G1 циклины.
G1/S циклины (Cln1 и Cln2 у S. cerevisiae, циклин E у позвоночных) достигает максимальной концентрации в поздней G1‑фазе и падает в S‑фазе.
G1/S cyclin–Cdk комплекс запускает начало репликации ДНК выключая различные системы подавляющие S-phase Cdk в G1‑фазе G1/S циклины также инициируют дупликацию центросом у позвоночных, образование веретенного тела у дрожжей. Падение уровня G1/S сопровождается увеличением концентрации S циклинов (Clb5, Clb6 у Sc и циклин A у позвоночных), который образует S циклин-Cdk комплекс который напрямую стимулирует ДНК репликацию. Уровень S циклина остается высоким в течении всей S, G2‑фаз и начала митоза, где помогает началу митозу в некоторых клетках.
М-циклины (Clb1,2,3 и 4 у Sc, циклин B у позвоночных) появляется последним. Его концентрация увеличивается, когда клетка переходит к митозу и достигает максимума в метафазе. М-циклин-Cdk‑комплекс включает сборку веретена деления и выравнивание сестринских хроматид. Его разрушение в анафазе приводит к выходу из митоза и цитокиезу. G1 циклины (Cln3 у Sc и циклин D у позвоночных) помогает координировать клеточный рост с входом в новый клеточный цикл. Они необычны, так как их концентрация не меняется от фазы клеточного цикла, а меняется в ответ на внешние регуляторные сигналы роста.
Программируемая клеточная гибель
В 1972 г. Керр с соавт. опубликовали статью, в которой авторы представили морфологические доказательства существования отличающегося от некроза особого вида гибели клеток, которую они назвали «апоптоз». Авторы сообщили, что структурные изменения клеток при апоптозе проходят две стадии:
1-я – образование апоптозных тел,
2-я – их фагоцитоз и разрушение другими клетками.
Причины гибели, процессы морфологического и биохимического характера развития клеточной смерти могут быть различными. Но все же их можно четко разделить на две категории:
1. Некроз (от греч. пеkrosis – омертвление) и
2. Апоптоз (от греч. корней, означающих «отпадение» или «распадение»), который часто называют программируемой клеточной смертью (ПКС) или даже клеточным самоубийством (рис. 354).
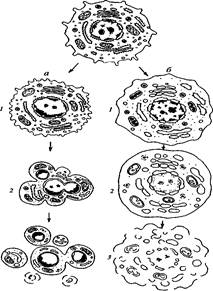
Два пути клеточной гибели
а – апоптоз (профаммированная клеточная смерть): / – специфическое сжатие клетки и конденсация хроматина, 2 – фрагментация ядра, 3 – фрагментация тела клетки на ряд апоптических телец; б – некроз: / – набухание клетки, вакуолярных компонентов, конденсация хроматина (кариорексис), 2 – дальнейшее набухание мембранных органоидов, лизис хроматина ядра (кариолизис), 3 – разрыв мембранных компонентов клетки – лизис клетки
НЕКРОЗ
Н. является наиболее частой неспецефической формой гибели клеток. Он может быть вызван тяжелыми повреждениями клетки в результате прямой травмы, радиации, влияния токсических агентов, вследствие гипоксии, лизиса клетки, опосредованного комплементом и т.д.
Некротический процесс проходит ряд стадий:
1) паранекроз – подобные некротическим, но обратимые изменения;
2) некробиоз – необратимые дистрофические изменения, характеризующиеся преобладанием катаболических реакций над анаболическими;
3) смерть клетки, время наступления которой установить трудно;
4) аутолиз – разложение мертвого субстрата под действием гидролитических ферментов погибших клеток и макрофагов. В морфологическом выражении некроз равнозначен аутолизу.
АПОПТОЗ.
Несмотря на огромное количество работ, согласованного и точного определения понятия «апоптоз» нет.
Алоптоз обычно характеризовали как особую форму гибели клетки, отличную от некроза по морфологическим, биохимическим, молекулярно-генетическим и другим признакам.
А. – это гибель клетки, вызываемая внутренними или внешними сигналами, которые сами по себе не являются токсичными или деструктивными. А. – это активный процесс, требующий затрат энергии, транскрипции генов и синтеза белка de novo.
Обнаружено значительное количество агентов, вызывающих апоптоз этих клеток, помимо облучения и глюкокортикоидов:
- ионофоры Са2+
- аденозин
- циклический АМФ
- трибутилтин
- АТФ
- гипертермия
Изучение кинетики деградации ДНК в лимфоидных клетках in vivo и in vitro показало:
- первые отчетливые признаки распада появляются, как правило, спустя более 1 ч после воздействия, чаще к концу 2‑го часа.
- Межнуклеосомная фрагментация продолжается в течение нескольких часов и заканчивается в основном через 6, реже 12 ч после воздействия.
Сразу же от момента появления деградации при анализе обнаруживается большое количество мелких фрагментов ДНК, причем соотношение между крупными и мелкими фрагментами в ходе апоптоза значительно не меняется.
Применение ингибиторов синтеза АТФ, белка и транскрипции генов замедляет процесс апоптоза. Такой зависимости в случае Н. нет
Как видно из сравнения определений некроза и апоптоза, между двумя видами гибели клетки имеется как сходство, так и существенные различия.
| Характеристика | Некроз | Апоптоз |
| функционально | необратимым прекращением ее жизнедеятельности; | необратимым прекращением ее жизнедеятельности; |
| морфологически | нарушением целостности мембран, изменением ядра (пикноз, рексис, лизис), цитоплазмы (отек), разрушением клетки; | потерей микроворсинок и межклеточных контактов, конденсацией хроматина и цитоплазмы, уменьшением объема клетки (сморщиванием), образованием пузырьков из плазматической мембраны, фрагментацией клетки и образованием апоптозных телец; |
| биохимически | нарушением выработки энергии, коагуляцией, гидролитическим расщеплением белков, нуклеиновых кислот, липидов; | гидролизом белков цитоплазмы и межнуклеосомным распадом ДНК; |
| генетически | – потерей генетической информации; и завершающейся ее аутолизом или гетеролизом с воспалительной реакцией. | структурно-функциональной перестройкой генетического аппарата и завершающийся ее поглощением макрофагами и(или) другими клетками без воспалительной реакции. |
Клеточная смерть регулируется межклеточными взаимодействиями различным образом. Множество клеток многоклеточного организма нуждается в сигналах с тем, чтобы оставаться живыми. В отсутствие таких сигналов или трофических факторов в клетках развивается программа «самоубийства» или программируемой смерти. Например, клетки культуры нейронов погибают при отсутствии фактора роста нейронов (NGF), клетки простаты гибнут в отсутствие андрогенов семенника, клетки молочной железы – при падении уровня гормона прогестерона и т.д. В то же время клетки могут получать сигналы, которые в клетках-мишенях запускают процессы, приводящие к гибели по типу апоптоза. Так, гидрокортизон вызывает гибель лимфоцитов, а глютамат – нервных клеток в культуре ткани, фактор некроза опухоли (TNF) вызывает гибель самых различных клеток. Тироксин (гормон щитовидной железы) вызывает апоптоз клеток хвоста головастиков. Кроме этого существуют ситуации, когда апоптическая гибель клетки вызывается внешними факторами, например радиацией.
Понятие «апоптоз» было введено при изучении гибели части клеток печени при неполной перевязке портальной вены. При этом наблюдается своеобразная картина клеточной смерти, которая затрагивает лишь отдельные клетки в паренхиме печени.
Процесс начинается с того, что соседние клетки теряют контакты, они как бы сморщиваются (первоначальное название этой формы гибели shrinkage necrosis – некроз сжатием клетки), в ядрах по их периферии происходит специфическая конденсация хроматина, затем ядро фрагментируется на отдельные части, вслед за этим сама клетка фрагментируется на отдельные тельца, отграниченные плазматической мембраной, – апоптические тельца.
Апоптоз – процесс, приводящий не к лизису, не к растворению клетки, а к ее фрагментации, распаду. Судьба апоптических телец тоже необычна: они фагоцитируются макрофагами или даже нормальными соседними клетками. При этом не развивается воспалительная реакция.
Важно отметить, что во всех случаях апоптоза – во время ли эмбрионального развития, во взрослом ли организме, в норме или при патологических процессах – морфология процесса гибели клеток очень сходна. Это может говорить об общности процессов апоптоза в разных организмах и в разных органах.
Исследования на разных объектах показали, что апоптоз есть результат реализации генетически запрограммированной клеточной гибели. Первые доказательства наличия генетической программы клеточной смерти (ПКС) были получены при изучении развития нематоды Caenorhabditis elegans. Этот червь развивается всего за трое суток, и его малые размеры позволяют проследить за судьбой всех его клеток, начиная с ранних этапов дробления до половозрелого организма.
Оказалось, что при развитии Caenorhabditis elegans образуется всего 1090 клеток, из которых часть нервных клеток в количестве 131 штуки спонтанно погибает путем апоптоза и в организме остается 959 клеток. Были обнаружены мутанты, у которых процесс элиминации 131 клетки был нарушен. Были выявлены два гена сеd‑3 и сеd‑4, продукты которых вызывают апоптоз 131 клетки. Если у мутантных Caenorhabditis elegans эти гены отсутствуют или изменены, то апоптоз не наступает и взрослый организм состоит из 1090 клеток. Был найден и другой ген – сеd‑9, который является супрессором апоптоза: при мутации сеd‑9 все 1090 клеток погибают. Аналог этого гена был обнаружен у человека: ген bcl‑2 также является супрессором апоптоза различных клеток. Оказалось, что оба белка, кодируемые этими генами, – Сеd‑9 и Вс1–2, имеют один трансмембранный домен и локализуются во внешней мембране митохондрий, ядер и эндоплазматического ретикулума.
Система развития апоптоза оказалась очень сходной у нематоды и позвоночных животных, она состоит из трех звеньев: регулятора, адаптера и эффектора. У Caenorhabditis elegans регулятором является Сеd‑9, который блокирует адаптерный белок Сеd‑4, который в свою очередь не активирует эффекторный белок Сеd‑3, протеазу, которая действует на белки цитоскелета и ядра (табл. 16).
Табл. 16. Развитие программируемой клеточной смерти (апоптоза)
| Объект | Регулятор | Адаптер | Эффектор | Результат | Результат |
| Caenorhabditis elegans | Ced‑9 ──┤ | Ced‑4 ─→ | Ced‑3 ─→ | ПКС | |
| Позвоночные | Bcl‑2 ──┤ | Apaf‑1 ─→ | Casp 9 ─→ | Casp 3 ─→ | ПКС |
Знак ──┤ – торможение процесса‚ знак ─→ – стимуляцию процесса
У позвоночных система ПКС более сложная. Здесь регулятором является белок Вс1–2, который ингибирует адаптерный белок Apaf‑1, стимулирующий каскад активации специальных протеиназ – каспаз.
Ферменты – участники процесса апоптоза
Таким образом,
- раз начавшись в клетке, такая деградация быстро протекает «до конца»;
- в апоптоз вступают не все клетки сразу или в короткий промежуток времени, а постепенно;
- разрывы ДНК происходят по линкерной (межнуклеосомной) ДНК;
- деградацию осуществляют эндо-, но не экзонуклеазы, и эти эндонуклеазы активируются или получают доступ к ДНК не в результате непосредственного взаимодействия с агентом, вызывающим апоптоз, а опосредованно, так как от момента контакта клеток с таким агентом до начала развития деградации проходит довольно значительное время, и, следовательно, фрагментация ДНК не является первой характерной «апоптотической» реакцией клетки на молекулярном уровне. В самом деле, если бы деградация запускалась в результате непосредственного взаимодействия эндонуклеаз или хроматина с агентом, то в случае, например, действия ионизирующей радиации апоптоз происходил бы быстро и одновременно почти во всех клетках.
- Исходя из этих заключений, расшифровка молекулярного механизма развития апоптоза «сосредоточилась» на идентификации эндонуклеаз(ы), осуществляющих фрагментацию ДНК, и механизмов, активирующих эндонуклеазы.
ЭндонуклеазыПохожие работы
... Циклины являются специфическими активаторами семейства циклин-зависимых протеинкиназ (CDK) (CDK - cyclin-dependent kinases) - ключевых участников индукции транскрипции генов, контролирующих клеточный цикл. Активация индивидуальной CDK происходит после ее взаимодействия со специфическим циклином, и образование этого комплекса становится возможным после достижения циклином критической концентрации. ...
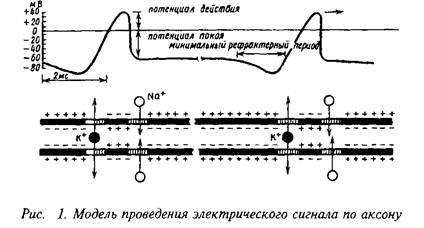
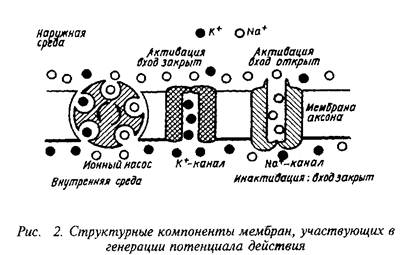
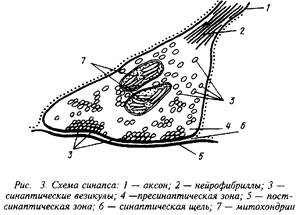
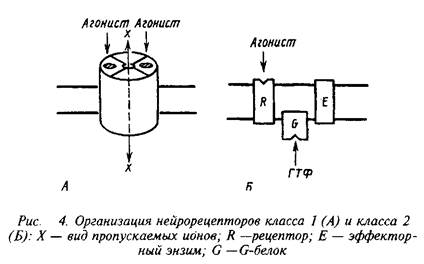
... роль нейрорецепторов сводится к созданию специфических информационных входов, организующих единый функциональный ансамбль нейронов. Именно совокупность рецепторов определяет лицо клетки и ее реакции на поступление разнообразных химических сигналов. Молекулярные механизмы, лежащие в основе модуляции эффективности синаптической передачи, в которых важную роль играют рецепторные процессы, имеют ...
... формированию представления о фундаментальных достижениях биологической химии в изучении гормональной регуляции процессов, протекающих в организме человека. Учебный курс «Молекулярные механизмы гормональной регуляции» должен в возможно максимальной степени отразить современное состояние и уровень развития биологической химии. Этот курс занимает важное место в системе подготовки биохимиков, ...
... произошли изменения не менее чем в 6 – 10 генетических факторах (теория многоступенчатого канцерогенеза). Изменения в пределах всего одной копии или аллеля протоонкогена достаточны для превращения его в онкоген с возросшей стимулирующей пролиферацию активностью, т.е. онкогены можно рассматривать как доминантные трансформирующие гены. Антионкогены, напротив, проявляются рецессивным образом т.е. ...
0 комментариев