Навигация
Деградацию осуществляет ДНКаза I. Процесс активируется Са2+ и Мg2+ [1979] и подавляется Zn2+ [1984]
33158
знаков
3
таблицы
2
изображения
1. Деградацию осуществляет ДНКаза I. Процесс активируется Са2+ и Мg2+ [1979] и подавляется Zn2+ [1984].
Однако имеются факты, которые свидетельствуют против участия ДНКазы I в процессе фрагментации ДНК. Так известно, что этот фермент отсутствует в ядре, правда, этот аргумент не очень весомый, так как относительно небольшой размер его молекул, 31 кДа, в случае нарушения проницаемости ядерной мембраны делает участие ДНКазы I в деградации ДНК вполне реальным. Другое дело, что при обработке хроматина in vitro ДНКаза I вызывает разрывы не только в линкерной части, но и в нуклеосомной ДНК.
2. Другой эндонуклеазой, рассматриваемой в качестве основного фермента деградации ДНК, является эндонуклеаза II [Барри 1993]. Эта нуклеаза при обработке ядер и хроматина осуществляет межнуклеосомную фрагментацию ДНК. Несмотря на то, что его активность не зависит от ионов двухвалентных металлов, вопрос об участии эндонуклеазы II в деградации ДНК не снят до сих пор, поскольку фермент не только находится в лизосомах, но и выделяется из ядер клеток.
3. эндонуклеаза с молекулярной массой 18 кДа. Этот фермент был выделен из ядер погибающих путем апоптоза тимоцитов крыс [Гайдо, 1991]. Она отсутствовала в нормальных тимоцитах. Активность фермента проявляется в нейтральной среде и зависит от Са2+ и Мg2+.
4. γ-нуклеаза с молекулярной массой 31 кДа, имеющая «классическую» зависимость от ионов Са, Мg и Zn. Активность этого фермента повышалась в ядрах тимоцитов крыс, обработанных глюкокортикоидами [1994].
5. эндонуклеаза с молекулярной массой 22,7 кДа фермент, активность которого проявляется в ядрах тимоцитов крыс только после действия глюкокортикоидов и подавляется теми же ингибиторами, что и межнуклеосомная деградация ДНК [1993].
Каспазы
Каспазы – цистеиновые протеазы, которые расщепляют белки по аспарагиновой кислоте. В клетке каспазы синтезируются в форме латентных предшественников – прокаспаз. Существуют инициирующие и эффекторные каспазы. Инициирующие каспазы активируют латентные формы эффекторных каспаз. Субстратами для действия активированных каспаз служат более 60 различных белков. Это, например, киназа фокальных адгезионных структур, инактивация которой приводит к отделению апоптических клеток от соседей; это ламины, которые при действии каспаз разбираются; это цитоскелетные белки (промежуточные филаменты, актин, гельзолин), инактивация которых приводит к изменению формы клетки и к появлению на ее поверхности пузырей, которые дают начало апоптическим тельцам; это активируемая протеаза САD, которая расщепляет ДНК на олигонуклеотидные нуклео-сомные фрагменты; это ферменты репарации ДНК, подавление которых предотвращает восстановление структуры ДНК, и многие другие.
Одним из примеров разворачивания апоптозного ответа может являться реакция клетки на отсутствие сигнала от необходимого трофического фактора, например фактора роста нервов (NGF), или андрогена.
В цитоплазме клеток в присутствии трофических факторов находится в неактивной форме еще один участник реакции – фосфорилированный белок Ваd. В отсутствие трофического фактора этот белок дефосфорилируется и связывается с белком Вс1–2 на внешней митохондриальной мембране и этим ингибирует его антиапоптозные свойства. После этого активируется мембранный проапоптический белок Вах, открывая путь ионам, входящим в митохондрию. В это же время из митохондрий через образовавшиеся в мембране поры в цитоплазму выходит цитохром с, который связывается с адаптерным белком Араf‑1, который в свою очередь активирует прокаспазу 9. Активированная каспаза 9 запускает каскад других прокаспаз, в том числе каспазу 3, которые, будучи протеиназами, начинают переваривать мешенные белки (ламины, белки цитоскелета и др.), что вызывает апоптическую смерть клетки, ее распад на части, на апоптические тельца.
Апоптические тельца, окруженные плазматической мембраной разрушенной клетки, привлекают отдельные макрофаги, которые их поглощают и переваривают с помощью своих лизосом. Макрофаги не реагируют на соседние нормальные клетки, но узнают апоптические. Это связано с тем, что при апоптозе нарушается асимметрия плазматической мембраны и на ее поверхности появляется фосфатидилсерин, негативно заряженный фосфолипид, который в норме располагается в цитозольной части билипидной плазматической мембраны. Таким образом, путем избирательного фагоцитоза ткани как бы очищаются от погибших апоптозных клеток.
Как указывалось выше, апоптоз может быть вызван целым рядом внешних факторов, таких как радиация, действие некоторых токсинов, ингибиторов клеточного метаболизма. Необратимые повреждения ДНК вызывают апоптоз. Это связано с тем, что накапливающийся транскрипционный фактор – белок р53, не только активирует белок р21, который ингибирует зависящую от циклина киназу и останавливает клеточный цикл в G1- или G2‑фазе, но и активирует экспрессию гена bax, продукт которого запускает апоптоз.Наличие контрольных точек в клеточном цикле необходимо для определения завершения его каждой фазы. Остановка клеточного цикла происходит при повреждении ДНК в G1 периоде, при неполной репликации ДНК в S‑фазе, при повреждении ДНК в G2‑периоде и при нарушении связи веретена деления с хромосомами.
Одним из контрольных пунктов в клеточном цикле является собственно митоз, который не переходит в анафазу при неправильной сборке веретена и при отсутствии полных связей микротрубочек с кинетохорами. В этом случае не происходит активации АРС-комплекса, не происходит деградации когезинов, соединяющих сестринские хрома-тиды, и деградации митотических циклинов, что необходимо для перехода в анафазу.
Повреждения ДНК препятствуют вхождению клеток в S‑период или в митоз. Если эти повреждения не катастрофические и могут быть восстановлены за счет репаративного синтеза ДНК, то блок клеточного цикла снимается, и цикл доходит до своего завершения. Если же повреждения ДНК значительные, то каким-то образом происходят стабилизация и накопление белка р53, концентрация которого в норме очень низкая из-за его нестабильности. Белок р53 является одним из факторов транскрипции, который стимулирует синтез белка р21, являющегося ингибитором комплекса СDК-циклин. Это приводит к тому, что клеточный цикл останавливается на стадии G1или G2. При блоке в G1‑периоде клетка с повреждением ДНК не вступает в S‑фазу, так как это могло бы привести к появлению мутантных клеток, среди которых могут быть и опухолевые клетки. Блокада в G2‑периоде также предотвращает процесс митоза клеток с повреждениями ДНК. Такие клетки, с блокированным клеточным циклом, в дальнейшем погибают путем апоптоза, программированной клеточной гибели (рис. 353).
При мутациях, приводящих к потере генов белка р53, или при их изменениях, блокады клеточного цикла не происходит, клетки вступают в митоз, что приводит к появлению мутантных клеток, большая часть из которых нежизнеспособна, другая – дает начало злокачественным клеткам.Избирательные повреждения митохондрий, при которых в цитоплазму высвобождается цитохром с, также являются частой причиной развития апоптоза. Особенно митохондрии и другие клеточные компоненты страдают при образовании токсически активных форм кислорода (АТК), под действием которых во внутренней мембране митохондрий образуются неспецифические каналы с высокой проницаемостью для ионов, в результате чего матрикс митохондрий набухает, а внешняя мембрана разрывается. При этом растворенные в межмембранном пространстве белки вместе с цитохромом с выходят в цитоплазму. Среди освободившихся белков есть факторы, активирующие апоптоз, и прокаспаза 9.
Многие токсины (рицин, дифтерийный токсин и др.), а также антиметаболиты могут вызывать гибель клеток путем апоптоза. При нарушении синтеза белка в эндоплазматическом ретикулуме в развитии апоптоза участвует локализованная там прокаспаза 12, которая активирует ряд других каспаз, и в том числе каспазу 3.
Элиминация – удаление отдельных клеток путем апоптоза, наблюдается и у растений. Здесь апоптоз включает в себя, так же как у животных клеток, фазу индукции, эффекторную фазу и фазу деградации. Морфология гибели клеток растений сходна с изменениями клеток животных: конденсация хроматина и фрагментация ядра, олигонуклеотидная деградация ДНК, сжатие протопласта, его дробление на везикулы, разрыв плазмодесм и т.д. Однако везикулы протопласта разрушаются гидролазами самих везикул, так как у растений нет клеток, аналогичных фагоцитам. Так, ПКС происходит при росте клеток корневого чехлика, при формировании перфораций у листьев, при образовании ксилемы и флоэмы. Опадание листьев связано с избирательной гибелью клеток определенной зоны черенка.
Биологическая роль апоптоза, или программированной смерти клеток, очень велика: это удаление отработавших свое или ненужных на данном этапе развития клеток, а также удаление измененных или патологических клеток, особенно мутантных или зараженных вирусами.
Итак, для того чтобы клетки в многоклеточном организме существовали, нужны сигналы на их выживание – трофические факторы, сигнальные молекулы. Эти сигналы могут быть переданы на расстояние и уловлены соответствующими рецепторными молекулами на клетках-мишенях (гормональная, эндокринная сигнализация), это может быть паракринная связь, когда сигнал передается на соседнюю клетку (например, передача нейромедиатора). При отсутствии таких трофических факторов реализуется программа апоптоза. В то же время апоптоз может вызываться сигнальными молекулами, например при резорбции хвоста головастиков под действием тироксина. Кроме того, действие ряда токсинов, влияющих на отдельные звенья метаболизма клетки, также может стать причиной клеточной гибели посредством апоптоза.
Апоптоз в патогенеза заболеваний
1. В иммунной системе
2. ОНКОЛОГИЧЕСКИЕ ЗАБОЛЕВАНИЯ
3. ВИРУСНАЯ ИНФЕКЦИЯ (индуцирующие апоптоз: в. иммунодефицита человека‚ в. анемии циплят; ингибирующие апоптоз: цитомегаловирус‚ в. Эпштейна-Барр‚ в. герпеса)
4. А. и НЕЙРОНЫ КОРЫ ГОЛОВНОГО МОЗГА
ПРИНЦИПЫ КОРРЕКЦИИ АПОПТОЗА КЛЕТКИ
Открытие регулируемого процесса гибели клетки – апоптоза–позволило определенным образом воздействовать на его отдельные этапы с целью регуляции или коррекции.
Биохимические процессы развития апоптоза можно гипотетически разделить на несколько этапов:
- действие фактора, вызывающего апоптоз;
- передача сигнала с рецепторной молекулы в клеточное ядро;
- активация апоптозспецифических генов;
- синтез апоптозспецифических белков
- активация эндонуклеаз
- фрагментацию ДНК (рис. 2.4).
В настоящее время считают, что если клетка погибает путем апоптоза, то подразумевается возможность терапевтического вмешательства, если вследствие некроза, то такое вмешательство невозможно. На основе знаний регуляции запрограммированной гибели клетки используется широкий ряд препаратов с целью воздействия на этот процесс в различных типах клеток.
Так, сведения о рецепторопосредованной регуляции апоптоза клеток учитывают при лечении гормонзависимых опухолей.
- Андрогенблокирующую терапию назначают при раке предстательной железы.
- Рак молочной железы часто подвергается регрессии при использовании антагонистов эстрогеновых рецепторов.
- Информация о биохимических сигналпередающих путях регуляции апоптоза позволяет эффективно применять антиоксидантную терапию, препараты, регулирующие концентрацию кальция, активаторы или ингибиторы различных протеинкиназ и т.д. с целью коррекции апоптоза в различных типах клеток.
Осознание роли апоптоза в гибели клеток интенсифицировало поиск фармакологических воздействий, защищающих клетки от апоптоза.
- Активно изучаются ингибиторы специфических протеаз в качестве фармакологических агентов. Это, как правило, три- или тетрапептиды, содержащие аспарагиновую кислоту (Асп). Использование таких протеаз в терапевтических целях ограничено их низкой способностью проникать в клетку. Однако, несмотря на это, в экспериментах in vivo успешно применяется Z-VAD-FMK [N‑бензилоксикарбонил-Вал-Ала-Асп(ОМе) – фторметилкетон] – ингибитор ICE‑подобных протеаз широкого спектра действия для снижения зоны инфаркта при моделировании инсульта.
- В ближайшие годы можно ожидать появления новых лекарственных средств для лечения и предупреждения различных заболеваний, основу действия которых будет составлять принцип регуляции процессов апоптоза.
Наиболее эффективны для коррекции апоптоза подходы, связанные с регуляцией апоптозспецифических генов. Эти подходы лежат в основе генной терапии – одного из перспективных направлений лечения больных с заболеваниями, вызванными нарушением функционирования отдельных генов.
Принципы генной терапии включают следующие этапы:
• идентификация последовательности ДНК, которая будет подвергаться лечению;
• определение типа клеток, в которых будет проводиться лечение;
• защита ДНК от гидролиза эндонуклеазами;
• транспорт ДНК в клетку (ядро).
Геннотерапевтические подходы позволяют
- усиливать работу отдельных генов (трансформация генов, ингибирующих апоптоз, например гена bcl‑2),
- ослаблять их экспрессию. Для селективного ингибирования экспрессии генов в настоящее время используют технику антисмысловых олигонуклеотидов (антисенсов). Использование антисенсов снижает синтез определенных белков, что влияет на регуляцию процесса апоптоза.
Механизм действия антисенсов активно изучается. В некоторых случаях короткие (13–17 оснований) антисмысловые олигонуклеотиды, имеющие последовательности, комплементарные нуклеотидным последовательностям матричной РНК (мРНК) отдельных белков, могут эффективно блокировать генетическую информацию на стадии, предшествующей транскрипции (рис. 2.5). Данные олигонуклеотиды, связываясь с ДНК, формируют триплетную спиральную структуру. Такое связывание может быть необратимым или вызывать селективное выщепление триплетного комплекса, что в итоге приводит к ингибированию экспрессии гена и гибели клетки. В других случаях происходит комплементарное связывание антисенса с мРНК, что вызывает нарушение трансляции и снижение концентрации соответствующего белка.
Триплетный комплекс
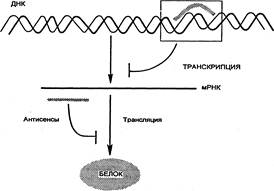
Рис. Регуляция экспрессии генов антисмысловыми олигонуклеотидами.
В настоящее время убедительно показано, что технология с использованием антисенсов имеет большое значение для регуляции отдельных генов в культуре клеток. Успешное подавление гена bcl‑2 в экспериментах на культурах клеток пробуждает надежду на применение в будущем антисенсов для лечения больных раком. Во многих экспериментах in vitro показано, что антисенсы вызывают ингибирование пролиферации и дифференцировки клеток. Такой результат подтверждает перспективы терапевтического использования данной технологии.
Похожие работы
... Циклины являются специфическими активаторами семейства циклин-зависимых протеинкиназ (CDK) (CDK - cyclin-dependent kinases) - ключевых участников индукции транскрипции генов, контролирующих клеточный цикл. Активация индивидуальной CDK происходит после ее взаимодействия со специфическим циклином, и образование этого комплекса становится возможным после достижения циклином критической концентрации. ...
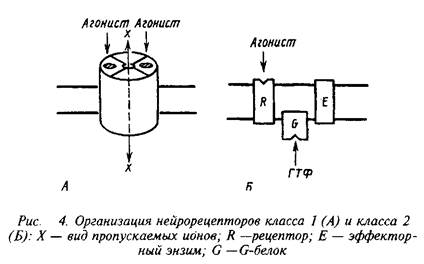
... роль нейрорецепторов сводится к созданию специфических информационных входов, организующих единый функциональный ансамбль нейронов. Именно совокупность рецепторов определяет лицо клетки и ее реакции на поступление разнообразных химических сигналов. Молекулярные механизмы, лежащие в основе модуляции эффективности синаптической передачи, в которых важную роль играют рецепторные процессы, имеют ...
... формированию представления о фундаментальных достижениях биологической химии в изучении гормональной регуляции процессов, протекающих в организме человека. Учебный курс «Молекулярные механизмы гормональной регуляции» должен в возможно максимальной степени отразить современное состояние и уровень развития биологической химии. Этот курс занимает важное место в системе подготовки биохимиков, ...
... произошли изменения не менее чем в 6 – 10 генетических факторах (теория многоступенчатого канцерогенеза). Изменения в пределах всего одной копии или аллеля протоонкогена достаточны для превращения его в онкоген с возросшей стимулирующей пролиферацию активностью, т.е. онкогены можно рассматривать как доминантные трансформирующие гены. Антионкогены, напротив, проявляются рецессивным образом т.е. ...
0 комментариев