Навигация
Законодательные и нормативные акты системы государственного регулирования лекарственного обеспечения
59268
знаков
0
таблиц
0
изображений
Законодательные и нормативные акты системы государственного регулирования
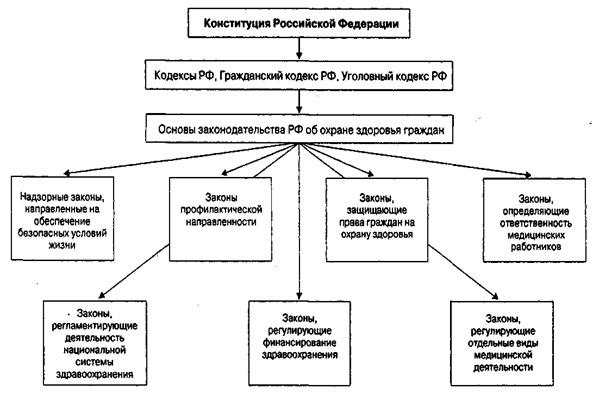
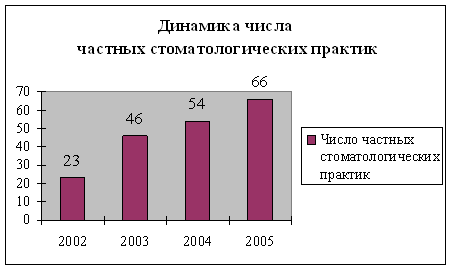
Лекарственное обеспечение населения и лечебно-профилактических учреждений является одной из приоритетных задач социальной политики российского государства на современном этапе. Для решения проблем охраны здоровья населения, выведения из кризисного состояния здравоохранения, медицинской промышленности и обеспечения граждан России жизненно необходимой лекарственной помощью за последнее время были приняты более 30 Указов Президента, постановлений и распоряжений Правительства Российской Федерации.
Основные направления, цели и задачи медицинской промышленности определены в программе "Развитие медицинской промышленности на 2005-2015 годы", разработанной Министерством экономики Российской Федерации, Министерством здравоохранения Российской Федерации, Научно-исследовательским институтом экономики медицинской промышленности.
Программа предусматривает развитие научно-технического и производственного потенциала медицинской промышленности с целью удовлетворения не менее чем на 70% потребностей здравоохранения и населения за счет отечественных диагностических средств, изделий медицинского назначения и медицинской техники надлежащего качества. Исходя из анализа экономической ситуации в стране и наличия финансовых средств, программа определяет тактические (на период 2005-2010 гг.) и стратегические (на период 2005-2015 гг.) цели, которые реализуются поэтапно.
Основными задачами программы являются:
¨ обеспечение в первоочередном порядке выпуска медицинской продукции для диагностики и лечения наиболее распространенных и социально значимых заболеваний (сердечнососудистых, сахарного диабета, онкологических, психических, инфекционных, включая СПИД, гепатиты, туберкулез), а также для охраны здоровья матери и ребенка по перечню жизненно необходимых и важнейших лекарственных средств;
¨ формирование эффективной системы обеспечения лекарственными средствами, изделиями медицинского назначения и медицинской техникой лечебно-профилактических учреждений и населения;
¨ разработка эффективного механизма привлечения финансовых средств коммерческих банков и других внебюджетных источников на развитие медицинской промышленности;
¨ обеспечение развития малых предприятий в медицинской промышленности;
¨ совершенствование системы мер по привлечению на конкурсной основе инвестиций в развитие медицинской промышленности;
¨ стимулирование лизинга для оснащения предприятий отрасли технологическим оборудованием и приборами;
¨ гармонизация нормативно-правовой документации со странами ЕС в области производства и реализации медицинской продукции;
¨ реформирование и реструктуризация предприятий, научных и проектно-конструкторских организаций в медицинской промышленности;
¨ осуществление мер государственной поддержки научно-исследовательских и опытно-конструкторских работ в медицинской промышленности, включая поддержку питомников лабораторных животных;
¨ создание современной системы организационно-экономического и информационно-аналитического обеспечения в сфере производства и реализации лекарственных средств, медицинской техники и изделий медицинского назначения, а также в области научно-исследовательских и опытно-конструкторских разработок;
¨ техническое перевооружение, реконструкция, расширение и строительство новых предприятий на базе прогрессивных технологий за счет всех источников финансирования;
¨ создание импортозамещающих производств;
¨ развитие экспортного потенциала;
¨ развитие производства субстанций лекарственных средств;
¨ развитие инфраструктуры, направленной на охрану окружающей среды, на действующих предприятиях медицинской промышленности;
¨ создание дополнительных рабочих мест за счет обеспечения ввода новых производственных мощностей;
¨ перепрофилирование ряда устаревших производств на выпуск новых, более эффективных видов медицинской продукции, внедрение новых и усовершенствованных технологических процессов, оборудования и средств автоматизации;
¨ концентрация усилий на приоритетных направлениях и повышение эффективности научных исследований по разработке и освоению современных лекарственных средств, медицинской техники и изделий медицинского назначения, эффективных видов сырья и материалов, новых и усовершенствованных технологических процессов, оборудования и средств автоматизации;
¨ разработка нормативно-правового и метрологического обеспечения медицинской промышленности.
Программой предусмотрены два этапа реализации.
Первый этап (2005-2007 гг.) – преодоление последствий финансового кризиса, повышение эффективности инвестиционных вложений за счет избирательного подхода к развитию производства готовых лекарственных средств и субстанций, пользующихся спросом на внутреннем и внешнем рынках медицинской продукции, осуществление модернизации и реформирование предприятий, обеспечение развития малых предприятий в медицинской промышленности, обеспечение выпуска импортозамещающей, конкурентоспособной продукции.
Второй этап (2007-2010 гг.) – экономический рост и реконструкция научно-производственного потенциала. На этом этапе осуществляется создание нового технологического фундамента для наращивания производства на основе высоких технологий. Вместе с тем, предусмотрено финансирование развития традиционных производств, не только на основе критерия перспективности производства (экспортный потенциал, тенденция к росту показателей эффективности), но и необходимости поддержки тех традиционных производств, которые могут ограничить потребительский импорт и характеризуются высокой добавленной стоимостью в мировых ценах.
Федеральным законом "О лекарственных средствах", принятым Государственной Думой 5 июня 2005 г., определены:
1. Государственное регулирование отношений в сфере обращения лекарственных средств, осуществляемое путем:
¨ государственной регистрации лекарственных средств;
¨ лицензирования деятельности в сфере обращения лекарственных средств;
¨ аттестации и сертификации специалистов, занятых в сфере обращения лекарственных средств;
¨ государственного контроля производства, изготовления, качества, эффективности, безопасности лекарственных средств.
2. Государственное регулирование обращения лекарственных средств осуществляется федеральным органом исполнительной власти и органами исполнительной власти субъектов Российской Федерации.
3. Государственная система контроля качества, эффективности, безопасности лекарственных средств включает:
¨ федеральный орган исполнительной власти и органы исполнительной власти субъектов Российской Федерации, в компетенцию которых входят осуществление государственного контроля качества, эффективности, безопасности лекарственных средств, надзор за фармацевтической деятельностью и иные действия в сфере обращения лекарственных средств;
¨ научно-исследовательские институты, лаборатории для разработки, исследований и осуществления государственного контроля качества, эффективности, безопасности лекарственных средств;
¨ экспертные советы по обращению лекарственных средств при Правительстве, действующие в соответствии с Положением об экспертных советах по обращению лекарственных средств, утверждаемым Правительством;
¨ этические советы, действующие при учреждениях здравоохранения в соответствии с Положением об этических советах, утверждаемым федеральным органом исполнительной власти в сфере здравоохранения;
¨ информационную систему, обеспечивающую субъекты обращения лекарственных средств необходимой информацией.
4. Полномочия по осуществлению контроля качества, эффективности, безопасности лекарственных средств возложены на федеральный орган контроля качества лекарственных средств, который является единственным федеральным органом исполнительной власти, ответственным за осуществление госконтроля. Эти полномочия не могут быть возложены на федеральный орган исполнительной власти, осуществляющий государственное управление промышленным производством лекарственных средств и медицинских изделий.
В Законе не установлены конкретные размеры таможенных пошлин. В целях защиты рынка и предприятий-производителей лекарственных средств на территории Российской Федерации правительство может вводить особые виды таможенных пошлин на импортные готовые лекарственные средства в соответствии с таможенным законодательством Российской Федерации.
В соответствии с Основами законодательства Российской Федерации о здравоохранении к ведению Министерства здравоохранения РФ относятся следующие вопросы:
¨ разрешение медицинского применения и регистрации лекарственных средств, медицинской техники и изделий медицинского назначения;
¨ утверждение государственных стандартов на лекарственные средства;
¨ сертификация и государственный контроль за качеством медицинской продукции;
¨ выдача лицензий на производство и реализацию лекарственных средств и медицинской техники.
Выполнение этих задач возложено на контрольно-разрешительную систему, функции управления которой осуществляет Управление государственного контроля лекарственных средств и медицинской техники.
Главной задачей контрольно-разрешительной системы является защита интересов потребителей от возможных негативных последствий применения лекарств, что может быть связано с недостаточной изученностью лекарственного средства на этапе его разрешения и внедрения в медицинскую практику, с выпуском предприятиями недоброкачественной продукции или с нарушениями условий и порядка его хранения и реализации. Это связано с тем, что уже в настоящее время в Государственный реестр лекарственных средств, зарегистрированных в Российской Федерации, включено более 10 000 наименований отечественных и зарубежных лекарственных средств. В соответствии с поставленными задачами контрольно-разрешительная система действует на двух уровнях: федеральном и региональном.
Основная задача на федеральном уровне – государственная регистрация лекарственных средств, изделий медицинского назначения и медицинской техники. На региональном уровне – контроль качества и сертификация лекарственных средств, изделий медицинского назначения и медицинской техники.
В качестве общественных экспертных организаций на федеральном уровне функционируют Фармакологический и Фармакопейный государственные комитеты, Федеральная комиссия по медицинским иммунобиологическим препаратам, Комитет по новой медицинской технике. Финансируются из федерального бюджета Государственный институт доклинической и клинической экспертизы лекарств (численность 200 человек), Государственный научно-исследовательский институт по стандартизации и контролю лекарственных средств (численность 158 человек) и Национальный орган по контролю медицинских иммунобиологических препаратов (ГИСК им. Тарасевича; численность 348 человек).
Управление государственного контроля лекарственных средств и медицинской техники и Государственный НИИ по стандартизации и контролю лекарственных средств осуществляют контроль качества лекарственных средств, выпускаемых всеми предприятиями Российской Федерации и ввозимых в Российскую Федерацию, инспектирование промышленных предприятий и аптечных учреждений в части соблюдения ими технологии производства, порядка контроля качества, хранения и реализации лекарственных средств, разработку инструктивно-методических документов по вопросам контроля качества лекарств и методическое руководство территориально-аналитическими лабораториями и отделами технического контроля предприятий.
На региональном уровне структура представлена территориальными контрольно-аналитическими лабораториями (центрами контроля качества лекарств), аккредитованными Минздравом России и зональными лабораториями государственного контроля препаратов крови, кровезаменителей и гемоконсервантов.
Для более строгого контроля установлены единые требования к процедурам регистрации лекарственных средств. Основные нормативные документы, входящие в состав инструкции о порядке регистрации, включают требования к субстанциям, к новым лекарственным средствам, к воспроизведенным препаратам, лицензии. В любом случае заявитель (фирма, отечественная или зарубежная) представляет документы в Министерство здравоохранения РФ, в Государственную инспекцию контроля качества лекарств и медицинской техники. Далее документация идет в Государственный Фармакологический комитет (Фармкомитет), в Государственный институт доклинической и клинической экспертизы и в Государственный фармакопейный комитет.
Важную роль в контрольно-разрешительной системе играет экспертный Комитет МИБП. Он рассматривает все вопросы, связанные с тактикой и стратегией применения МИБП. Все новые МИБП проходят через этот Комитет. Комитет состоит из трех секций, куда входят 80 ведущих экспертов в области бактериологии, вирусологии, иммунологии, эпидемиологии, инфекционных, паразитарных, аллергических болезней и другие специалисты по теоретическим, прикладным и клиническим вопросам МИБП. Заключения Комитета являются основанием для принятия решения о применении препаратов в медицинской практике или об изъятии устаревших препаратов.
В отличие от многих стран, в России существует система государственных испытаний, которые не зависят от разработчиков вакцин и проводятся под руководством контрольного института. Такая система оправдала себя, она крайне важна в условиях нестабильных хозяйственных и правовых отношений. Все это позволяет достаточно точно определить недостатки и преимущества испытуемого препарата. Подобные же испытания проходят и зарубежные препараты. По существующему положению на территории России нельзя применять импортные МИБП, не прошедшие регистрацию.
Система надзора за МИБП основана на принципах гарантии качества. Контроль за качеством МИБП не заканчивается регистрацией этого препарата, он продолжается на всех этапах производства в цехах и в отделах биологического и технологического контроля предприятий, в ГИСК им. Л.А. Тарасевича, который осуществляет сертификацию самих МИБП и их производство.
В 1997 г. Госстандарт России зарегистрировал самостоятельную систему сертификации МИБП. В целом по стране ежегодно осуществляется лабораторный контроль 5-10% серий всех вакцин и 20-40% серий препаратов календаря прививок и тест-систем для диагностики ВИЧ-инфекции и гепатитов В и С. В некоторых развитых странах контроль серий наиболее важных препаратов составляет 80–100%. Что касается диагностических препаратов, то в России их испытания проводятся только на стадии внедрения в практику, при пересмотре или продлении документации. В связи с эпизодическим контролем этой группы препаратов в РФ нет систематического мониторинга за качеством диагностических МИБП. Все это свидетельствует о необходимости усиления контроля за качеством диагностических препаратов.
В 1996 г. в России введен так называемый предреализационный контроль вакцин по сводным протоколам производства и контроля. Такие протоколы составляются на предприятиях по формам, рекомендованным ВОЗ, и направляются в Контрольный институт. Предприятие не имеет права отгружать вакцину потребителю без заключения ГИСК им. Л.А. Тарасевича. Такому предреализационному контролю подвергаются все серии вакцин календаря обязательных прививок.
Письменное заключение о результатах государственного предварительного контроля вместе с протоколом анализа Институт направляет предприятию-изготовителю, а копии в Управление. До получения результатов анализа от Института контролируемые и все последующие серии лекарственного средства реализации не подлежат.
Лекарственное средство снимается с режима предварительного контроля письменным разрешением Управления и переводится на последующий государственный контроль, если качество образцов препарата первых пяти серий отвечает всем требованиям нормативного документа.
В случае, если при проведении предварительного контроля Институтом были сделаны замечания к качеству лекарственного средства, то оно с предварительного контроля не снимается. Количество серий, досылаемых на повторный контроль, и проверяемые показатели качества определяются Институтом.
Последующему и выборочному контролю подвергаются серийно выпускаемые лекарственные средства. Номенклатура и периодичность отбора лекарственных средств устанавливаются планами-заданиями, утвержденными Управлением.
Отбор образцов на последующий и выборочный контроль на предприятиях-изготовителях проводится отделом технического контроля с участием представителя территориальной контрольно-аналитической лаборатории. Отбор образцов на аптечных базах (складах), в аптеках и других учреждениях, реализующих и использующих лекарственные средства, производится представителем территориальной контрольно-аналитической лаборатории, основанием для изъятия лекарственных средств служит предписание Управления Госконтроля. Отбор образцов может также проводиться представителями Института или других организаций, учреждений и предприятий по заданию Управления, а также при обследованиях предприятий-изготовителей, аптечных и лечебных учреждений или по сигналам учреждений здравоохранения и населения.
Образцы препаратов с сопроводительным письмом и актом отбора средней пробы направляются в Институт. Для инъекционных растворов и глазных капель должно дополнительно прилагаться письменное заключение территориальной контрольно-аналитической лаборатории с результатами проверки по показанию “Механические включения” в соответствии с действующими инструкциями.
В случае выявления несоответствия качества образцов лекарственных средств требованиям нормативного документа на стадии последующего контроля Институт направляет письменное заключение с протоколом анализа Управления предприятию-изготовителю и учреждению, представившему образцы. При положительных результатах анализов Институт уведомляет только учреждение, представившее образцы.
Арбитражному контролю подвергаются лекарственные средства в случае возникновения споров об их качестве между поставщиком и потребителем. Для проведения арбитражных анализов образцы препаратов направляются в Институт с сопроводительным письмом, актом отбора средней пробы, протоколом анализа по всем показателям нормативного документа и документом, подтверждающим отказ изготовителя (поставщика) удовлетворить претензию потребителя. Если согласование вопроса о качестве препарата между потребителем и изготовителем (поставщиком) не достигается в течение одного месяца, допускается представление документа, подтверждающего направление претензии изготовителю (поставщику) взамен его отказа удовлетворить претензию.
Если порядок последующего (выборочного) и арбитражного контроля для отечественных и зарубежных лекарственных средств однотипный, то в проведении предварительного контроля зарубежных препаратов имеются некоторые особенности. Так, предварительному контролю подлежат зарегистрированные лекарственные вещества (субстанции), поступившие от одной фирмы и предназначенные для изготовления готовых лекарственных форм отечественными предприятиями.
Разрешение на использование зарубежных субстанций в производстве отечественных лекарственных форм выдается предприятию Управлением при наличии сертификата анализа фирмы-производителя (поставщика) и после проверки их качества в Институте. Незарегистрированные лекарственные вещества после ввоза в страну в 10-дневный срок должны быть направлены на контроль в Институт. Разрешение на ввоз незарегистрированных лекарственных средств выдается Управлением.
Экспертиза нормативных документов осуществляется Институтом в процессе Государственного контроля, который включает в себя не только проверку качества лекарственных средств на их соответствие требованиям нормативного документа, но и независимую экспертизу самих документов, которые являются государственными стандартами и должны обеспечивать получение достоверной и достаточной информации о качестве анализируемых лекарственных средств.
Специалисты Института при осуществлении экспертизы проводят оценку уровня нормативного документа в сравнительном аспекте с требованиями передовых зарубежных фармакопей и нормативных документов на аналогичные препараты других стран и фирм. Это возможно только при наличии банка данных, накопленных Институтом за его многолетнюю практику. Такой подход к проведению экспертизы согласуется с международной практикой государственных контрольных институтов других стран.
При оценке качества лекарственных средств, как правило, используется комплекс физико-химических, биохимических и биологических методов, описанных в Фармакопее. Необходимость сочетания этих методов определяется видом лекарственной формы и ее принадлежностью к фармакологической группе. Так, например, при контроле препаратов инсулина используются практически все приведенные в Фармакопее методы.
Отдельного реестра по стандартным образцам (СО) лекарственных средств нет. В связи с тем, что на СО утверждается такой же нормативный документ и в том же самом порядке, как на лекарственные средства, то они включаются в общий государственный Реестр лекарственных средств с присвоением им номера госрегистрации.
Номенклатура лекарственных средств и задачи, определенные в Уставе института, определяют его структуру. В соответствии с указанными документами в институте сформировано 13 структурных подразделений, из них 8 являются ведущими лабораториями, осуществляющими контроль по закрепленной номенклатуре, 3 лаборатории – вспомогательные: лаборатория биохимии лекарственных средств, лаборатории микробиологии и фармакологии, которые осуществляют контроль отдельных показателей (стерильность, микробиологическая чистота, определение токсичности, пирогенности и биологической активности, ферментативной активности лекарственных средств).
Лаборатория научной организации государственного контроля лекарственных средств осуществляет все делопроизводство института, обеспечивает ведущие лаборатории нормативными документами и принимает участие в разработке нормативных актов, регламентирующих порядок деятельности контрольно-разрешительной системы. Лаборатория научной стандартизации лекарственных средств – рабочий орган Фармакопейного комитета. Сотрудники этой лаборатории являются секретарями специализированных комиссий.
Фармакопейный государственный комитетОсновным нормативным документом, регулирующим сегодня деятельность Фармакопейного комитета, является положение о Фармакопейном комитете, утвержденное в конце 1992 г. Согласно этому положению Фармакопейный комитет работает при Министерстве здравоохранения.
В комитет входят 12 комиссий, семь из которых химико-фармацевтические – каждой из них приданы права рассматривать документацию на препараты определенного профиля (например, фитопрепараты, гормональные средства, антибиотики и др.), а две – по иммунобиологическим препаратам. Кроме того, существуют две специализированные комиссии. Одна из них – фармакологическая, анализирует фармакологические методы определения и дает оценку документации с точки зрения фармакологии. Другая – микробиологическая, рассматривает микробиологические методы и оценивает документацию с точки зрения микробиологии. Еще две комиссии рассматривают иммунобиологические препараты: соответственно по противобактериальным препаратам и противовирусным препаратам с правом рассмотрения документации на аллергены.
В 1995 году в связи с подготовкой нового издания фармакопеи России создана комиссия по экспертизе документации на отечественные лекарственные средства. Таким образом, российские лекарственные средства оцениваются двумя комиссиями: одной из химико-фармацевтических и в обязательном порядке комиссией по экспертизе. Комиссии руководствуются пятью фармакопеями: российской, американской, британской, немецкой и фармакопеей ЕС.
Над комиссиями стоит высший орган – Президиум. Он рассматривает предложения комиссий и ставит окончательную точку в экспертизе документации, давая свое решение в форме рекомендации Министерству здравоохранения. Работает также группа экспертов, в составе которой более ста человек. Каждый из экспертов привлекается в случае необходимости.
Кроме того, необходимо отметить особое участие в работе Фармакопейного комитета лаборатории научной организации государственного контроля лекарственных средств, которая входит в структуру ГНИИСКЛС. Все сотрудники этой лаборатории – одновременно ученые секретари комиссий Фармакопейного комитета, а сама лаборатория – рабочий орган Фармакопейного комитета. Общее число участников экспертизы на отечественные и зарубежные средства в Фармакопейном комитете – около 300 человек.
Число членов комиссии колеблется от 9 до 13 человек. Президиум состоит из 5 названных единиц, назначенных приказом министра, 12-ти председателей комиссий и 3-х представителей других организаций с правом совещательного голоса: представителя Фармакологического комитета, представителя Управления государственного контроля, представителя Национального центра по контролю иммунобиологических препаратов. В семи комиссиях имеются сопредседатели, которые в определенных случаях тоже вводятся в состав президиума с правом решающего голоса.
Президиум собирается круглый год каждую среду при кворуме 50% его членов, но меньше 80% на заседаниях не бывает. При отсутствии председателя комиссии он пишет официальное уведомление, в котором назначает того, кто заместит его на текущем Президиуме в данный период. Этот человек во время отсутствия своего председателя возглавляет комиссию.
Принципиальная схема составления регистрационного досье остается незыблемой: в нем предусмотрены все элементы, необходимые Фармакопейному комитету в процессе экспертизы. Для каждой лекарственной формы на импортные ЛС существует определенный стандарт, разработанный самим Фармакопейным комитетом; для отечественных препаратов существует отраслевой стандарт по подготовке документов по стандартизации – временных фармакопейных статей или фармакопейных статей. Заявитель направляет документы по стандарту в Минздрав, а Минздрав в Фармакопейный комитет. С 1995 г. комитет вообще не имел дела с заявителем. Сроки прохождения документов лимитированы. Через три месяца (в них не входит время, за которое заявители отвечают на вопросы Фармакопейного комитета) после получения заявки Фармакопейным комитет должен вынести Министерству здравоохранения рекомендации по поводу регистрации.
Комиссия выносит свое предварительное решение. Затем его корректирует, редактирует и визирует ученый секретарь. Затем документ опять попадает в комиссию. Она принимает окончательное решение – выносит дело на президиум или нет, рекомендовать или не рекомендовать ЛС. Далее – президиум, после него председатель Фармакопейного комитета подписывает соответствующий протокол.
Институт доклинической и клинической экспертизы лекарств (ГИДКЭЛ)Основная задача Института – экспертиза всех материалов по доклиническим и клиническим исследованиям лекарственных средств для регистрации в Российской Федерации.
Институт доклинической и клинической экспертизы лекарств (ГИДКЭЛ) был создан на базе Российского государственного центра экспертизы лекарств и является его приемником. В институте работают следующие отделы: отдел фармацевтической экспертизы, отдел переносимости лекарственных средств, который объединил в себе отдел токсикологии, далее отдел организации контрольно-клинических испытаний, а также ряд специализированных отделов: экспертизы химико-терапевтических средств, сердечно-сосудистых лекарственных средств, иммунологических и аллергических, аллергологических средств, применяемых при заболеваниях центральной нервной системы и другие.
В своей работе ГИДКЭЛ руководствуется инструкциями Министерства здравоохранения РФ о порядке экспертизы клинических испытаний и регистрации зарубежных лекарств, о регистрации испытаниях отечественных лекарств, а также приказом об утверждении состава Фармакологического комитета и положением о нем. Это основные документы, в настоящее время они пересматриваются и дополняются в связи с возникновением новых вопросов и проблем. Будут учтены и четвертая фаза испытаний, и список безрецептурных (далее – ОТС) лекарств, разработанный институтом совместно с НИИ фармации. Последний список был издан очень давно.
В новые документы входит и такой аспект, как листовки-вкладыши с аннотацией средств. Это вызвано тем, что часто в средствах массовой информации рекламируются различные лекарства и пищевые добавки, которые не прошли испытания, не соответствуют действительности и вводят в заблуждение население. Это является нарушением закона о правах потребителя. Кроме того, в продажу поступают препараты, о которых никто не знает, они не имеют аннотаций, или прилагаемая инструкция не переведена на русский язык, или переведена, но неграмотно.
Вся документация поступает в Институт, где предварительно рассматривается экспертами. При этом учитывается ее количество, объем, качество исследований. Иногда количество документов составляет 10-12 томов, и даже больше. Если документов недостаточно, заявителю посылаются дополнительные вопросы. По существующему положению к клиническим испытаниям допускаются следующие препараты: новые оригинальные зарубежные и отечественные; уже известные, но с новыми показаниями; новые комбинации лекарственных средств; ЛС, уже хорошо известные, но поданные в более высоких дозах, чем зарегистрированные ранее. В сложных случаях для решения вопроса привлекаются специализированные комиссии Фармакологического комитета. И только после этого препарат поступает на клинические испытания.
По международным стандартам существует несколько клинических испытаний. Первая – начальный этап, проводимый на небольшой группе здоровых людей. Второй этап – исследования проводятся на больных людях, где оценивается эффективность и переносимость, уточняются показания и противопоказания, определяются оптимальные дозировки. В третьей фазе проводятся мультицентровые клинические испытания. В четвертой фазе проводятся пострегистрационные испытания.
После регистрации препарат широко используется в медицинской практике, что позволяет уточнить особенности его действия, которые не могли быть замечены в предыдущих трех фазах. При этом очень важных аспект – экономические показатели для использования. В предшествующие десятилетия четвертую фазу у нас в стране не практиковали.
Испытания выполняются в соответствии с протоколом, т.е. перед началом испытаний члены специализированных комиссий совместно со специалистами Института составляют единый протокол испытаний данного препарата. Все клиники работают в соответствии с этим протоколом. Поэтому в результате получаются унифицированные данные и практические отчеты, написанные в одном и том же ключе, что дает возможность более точно и полноценно оценить эффективность и безопасность исследуемого средства.
Институт совместно с Фармакологическим комитетом разработал специальную карту с вопросами и создал специализированную комиссию по оценке и сертификации клинических баз, только на которых будут проходить испытания.
Уровень испытаний настолько высок, что международные компании готовы заключать соглашения о проведении мультицентровых клинических испытаний. Клинические базы России сотрудничают с такими крупными фирмами, как "Merk sharp & Dome" (США), "Pfizer" (США), "Ron-Pulenk" и другими. В настоящее время при крупных лечебных центрах созданы комитеты по этике, которые дают одобрение на все клинические испытания учреждения.
В январе 1994 г. был подписан Меморандум о взаимопонимании между Управлением пищевых продуктов и лекарств Министерства здравоохранения и социального обеспечения США и Министерством здравоохранения и медицинской промышленности РФ и Госкомсанэпиднадзором РФ о сотрудничестве и обмене информацией о лекарствах и биологических препаратах и содействии их импорту. Одним из разделов этого меморандума предусматривается оптимизация требований при регистрации в России американских лекарств и биологических препаратов, которые производятся в США и допущены FDA к продаже в США.
Для регистрации американских препаратов фирма представляет такое же полное досье, как и во всех других случаях, проводится обязательная экспертиза этой документации в Фармакологическом и Фармакопейном комитетах, но могут не проводиться экспериментальные и клинические исследования препаратов, если эти данные подробно представлены в досье.
В настоящее время подобные соглашения заключены Минздравмедпромом с Францией и с Канадой. Ведутся переговоры о заключении соглашения со Швейцарией и странами, входящими в Европейский Союз. Это позволило увеличить количество регистрируемых в России лекарственных средств из указанных стран.
Увеличение регистрации и поступления на рынок России лекарств из стран с высокоразвитой фармацевтической промышленностью способствует сокращению регистрации большого потока поступающих в Россию лекарств (в основном дженериков) из стран с менее развитой фармакологической промышленностью и боле низкого качества (Индия, Индонезия, Бангладеш и др.)
В соответствии с новой инструкцией "О порядке экспертизы, клинических испытаний и регистрации лекарственных средств и субстанций", утвержденной 10 июля 2004 г., был введен новый порядок проведения и оплаты экспертизы документации при регистрации лекарственных средств ГИДКЭЛ, Фармакологическим, Фармакопейным комитетом и другими организациями. Теперь экспертиза проводится в соответствии с договорами, заключенными этими учреждениями с Управлением и оплачивается из средств Бюро по регистрации в соответствии с техническим заданием в порядке, установленном Управлением. Срок проведения экспертизы – 90 дней. Введение нового порядка позволило усилить контроль за сроком и качеством проведения экспертизы.
В 2003 году подготовлено новое издание Государственного реестра лекарственных средств, в которое вошло более 10 000 наименований лекарственных средств. Значительно переработано содержание Госреестра, в том числе впервые приведены коды анатомо-терапевтическо-химической классификации лекарственных средств, рекомендованной ВОЗ.
Впервые начата работа по созданию Государственного реестра инструкций по применению лекарственных средств, утвержденных Фармакологическим государственным комитетом.
Система контроля качестваВажнейшим этапом деятельности контрольно-разрешительной системы является контроль качества серийно выпускаемых лекарственных средств путем сравнения образцов продукции с утвержденным стандартом.
Система контроля качества серийно выпускаемых лекарственных средств действует в России в трех уровнях. На федеральном уровне эту систему представляют:
¨ Управление государственного контроля лекарственных средств и медицинской техники;
¨ ГосНИИ по стандартизации и контролю лекарственных средств и Центральная лаборатория государственного контроля и изучения качества препаратов, кровезаменителей и консервирующих растворов Гематологического научного центра РАМП, имеющие функции:
1. Проведения всех типов государственного контроля:
¨ предварительного – первых 5 серий препарата, впервые производимого данным предприятием или выпускаемого по измененной технологии, или переведенного Управлением на это вид контроля в связи с ухудшением его качества, а также импортных субстанций;
¨ выборочного последующего (любая серия препарата, изъятая со склада предприятия-изготовителя или с места хранения, или из аптеки) по планам-заданиям, утверждаемым Управлением;
¨ арбитражного;
¨ сертификации лекарственных средств.
2. Аттестации и рассылки государственных стандартных образцов для контроля качества лекарственных средств.
3. Участия в инспекционных проверках, организации и осуществления контроля за качеством лекарственных средств на предприятиях и в учреждениях, производящих, хранящих и реализующих лекарственные средства, независимо от их организационно-правовой формы.
Похожие работы
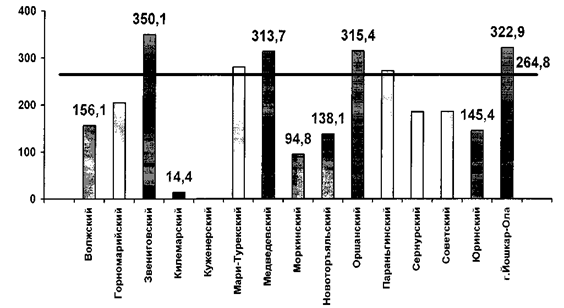
... процессов формирования и укрепления здоровья населения должные оказывать существенное влияние на совершенствование охраны здоровья населения Республики Марий Эл. 3. Основные направления совершенствования государственного регулирования здравоохранения в Республике Марий Эл 3.1. Разработка и реализация республиканских программ по охране и укреплению здоровья населения В «Основных ...
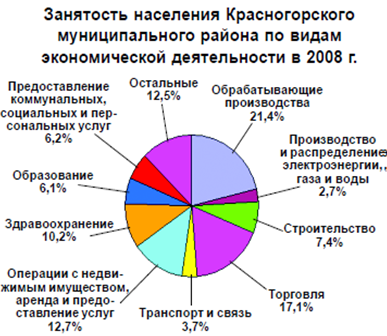
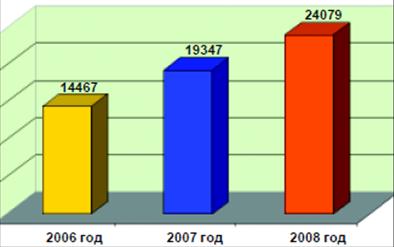
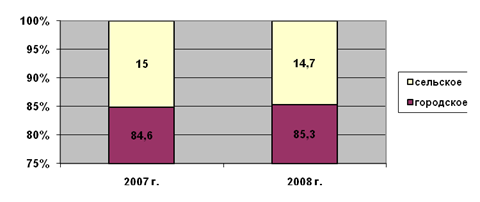
... ); 4. открытие отделения экстракорпоральной дотоксикации (искусственная почка) на базе КГБ №1. 3. Проектная часть. Разработка проектных мероприятий Основные направления совершенствования финансирования системы здравоохранения Красногорского района Московской области Обеспеченность финансовыми ресурсами государственных гарантий населению в сфере здравоохранения. В сложившейся ...
... множество конкретных целей, без осуществления которых не может быть достигнута главная цель. Эти конкретные цели неразрывно связаны с объектами государственного регулирования рынка стоматологических услуг Чувашской Республики. "Дерево целей" видоизменяется в зависимости от конкретных проблем, стоящих перед Чувашской Республикой. Конкретные цели можно сгруппировать по определенным блокам, ...
... с такой социальной политикой наше государство сможет пережить с минимальными социальными убытками мировой финансовый кризис, заставший врасплох и социальный сектор нашей страны. 1.3 Государственное регулирование социального развития в условиях мирового финансового кризиса После десяти лет непрерывного экономического роста и повышения благосостояния людей Россия столкнулась с серьезнейшими ...
0 комментариев