Навигация
Покровные стекла инкубируют с антивирусной сывороткой 1 ч при 37 °С, промывают и инкубируют еще 45 мин при 37 °С с соответствующим конъюгатом
50436
знаков
1
таблица
5
изображений
5. Покровные стекла инкубируют с антивирусной сывороткой 1 ч при 37 °С, промывают и инкубируют еще 45 мин при 37 °С с соответствующим конъюгатом.
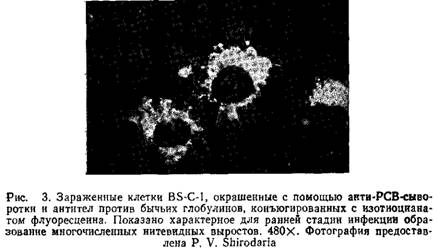
6. После промывки препараты заключают в забуференный глицерин и исследуют под флуоресцентным микроскопом. Рис.3 иллюстрирует разрешение, которое может быть получено при использовании данного метода.
Доказательствами специфичности иммунофлуоресценции могут служить:
1) отсутствие флуоресценции незараженных клеток BS-C-1;
2) отсутствие флуоресценции при использовании ЭТС вместо специфической с зараженными РСВ клетками;
3) отсутствие флуоресценции после истощения противовирусной сыворотки зараженными клетками.
Высокой чувствительностью обладает и иммунопероксидазный метод; его преимущество состоит в том, что он не требует использования микроскопа с ультрафиолетовой оптикой.
2.6 Твердофазный иммуноферментный анализELISA - еще один иммунологический метод быстрого обнаружения и количественного определения РСВ. Ниже приведена типичная методика.
1. Лунки планшета для иммунологических реакций покрывают зараженными или незараженными клетками, или РСВ, очищенным центрифугированием в градиенте. Это делается одним из следующих методов:
а) клетки выращивают в лунках планшета и через ряд заражают РСВ. Когда в лунках с зараженными клетками проявляется ЦПД, клетки промывают, фиксируют раствором, содержащим этанол и уксусную кислоту, и хранят при - 20 °С;
б) в лунки планшета вносят РСВ, очищенный центрифугированием в градиенте, и высушивают в течение ночи при 37°С.
Если имеются клетки, персистентно инфицированные РСВ, их можно использовать в качестве источника антигена вместо литически инфицированных клеток.
2. Перед началом анализа все лунки планшета покрывают 5% -ным раствором БСА в PBS. В каждый опыт следует включать негативный контроль и позитивный контроль. В лунки планшета вносят образцы вируса и инкубируют в течение ночи при 4°С. Планшет промывают и добавляют овечьи антитела против иммуноглобулинов мыши, конъюгированные с пероксидазой хрена в рекомендуемом разведении. Планшет инкубируют 1 ч при 37 °С, промывают, добавляют субстрат, о-фенилендиамин, и инкубируют при 37 °С еще 30 мин. Реакцию останавливают добавлением 4,5 М H2SO4 и учитывают результаты с помощью ридера "Мультискан".
Хиерхольцер и др. описали методику, позволяющую с помощью ELISA количественно выявлять белковые полосы после электрофореза и переноса белков на нитроцеллюлозные мембраны, так называемый метод "вестерн-блотинга". Преимущество этого метода состоит в том, что он не требует использования радиоактивных изотопов.
3. Аналитические методы 3.1 Радиоактивное лечение, радиоиммуноанализ и радиоиммунопреципитация 3.1.1 Введение радиоактивной метки в вирусные белки в зараженных РСВ клетках
1. Оптимальные условия для мечения вирусных белков достигаются при добавлении к зараженным клеткам через 18 ч после заражения актиномицина D в концентрации 2,5 мкг/мл.
Белки РСВ удается эффективно пометить смесью - Meтионина и - цистеина по следующей методике.
2. Через 2 ч после внесения актиномицина D культур ал ьную среду заменяют на среду, содержащую 50 мкКи/мл L--Meтионина и 25 мкКи/мл Ь--цистеина. у
3. Клетки инкубируют 16 ч при 37 °С или до проявления выраженного ЦПД.
4. Монослой промывают, солюбилизируют клетки и экстрагируют белки лизирующим буфером.
5. После инкубации в течение 2 ч при комнатной температуре лизат используют для электрофореза или хранят при - 20 °С.
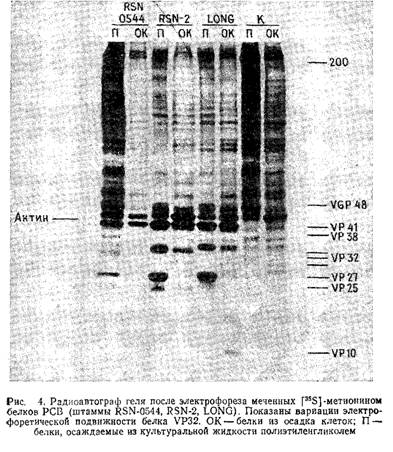
Рис.4 иллюстрирует результаты разделения меченных - метионином белков РСВ в пластине 10% -ного полиакриламидного геля. Вирусные гликопротеины можно пометить аналогичным методом, используя в качестве метки 50 мкКи/мл - глюкозамина или - маннозы. Альтернативный метод введения метки в гликопротеины оболочки РСВ заключается в йодировании поверхности зараженных вирусом клеток лактопероксидазным методом. Белки очищенного нуклеокапсида РСВ можно йодировать с помощью хлорамина Т.
Метод прямого RIA с использованием в качестве вторых антител меченных 1251 гамма-глобулинов сыворотки козы против глобулинов мыши, а в качестве субстрата - фиксированных метанолом персистентно инфицированных РСВ клеток описан Коутом и др. .
3.1.3 РадиоиммунопреципитацияРадиоиммунопреципитация проводится стандартным методом, описанным, например, Ферни и Герином и Уордом и др. Для РСВ применяют следующую методику.
1. "Монослойные культуры клеток в чашках Петри диаметром 50 мм заражают РСВ с множественностью инфекции более 1 БОЕ/кл. Через определенные промежутки времени после заражения проводят импульсное мечение. Для этого культуральную среду в чашках заменяют на 1 мл PBS, содержащего 30 мкКи - метионина и 25 мкКи - цистеина, и инкубируют чашки при 31 °С 1 ч.
2. Радиоактивный раствор сливают и монослой промывают охлажденным PBS.
3. Клетки снимают со стекла с помощью стерильной резиновой палочки и осаждают центрифугированием.
4. Клетки ресуспендируют в 200 мкл лизирующего буфера, инкубируют суспензию 30 мин в ледяной бане, затем интенсивно перемешивают.
5. Ядра и клеточный дебрис удаляют центрифугированием 5 мин при 10 000 g.
6. Лизат осветляют инкубацией с 50 мкл 10% -ной суспензии фиксированных формалином стафилококков или с 50 мкл суспензии покрытых белком-А гранул 30 мин при 0°С.
7. Стафилококки удаляют центрифугированием. К 50 мкл лизата добавляют 10 мкл гипериммунной сыворотки, перемешивают и инкубируют 3 ч во льду.
8. Добавляют 100 мкл суспензии стафилококков, перемешивают и инкубируют смесь 30 мин при комнатной температуре.
9. Иммунные комплексы осаждают центрифугированием 20 с при 10 000 g.
10. Осадок 3 раза промывают раствором, содержащим 500 мМ LiCl, 100 мМ трис-HCl, рН 8,5.
11. Осадок после промывки ресуспендируют в 50 мкл смеыг для диссоциации, кипятят 2 мин и либо немедленно наносят на гель для электрофореза, либо хранят при - 20 °С.
3.2 Метод выявления дефектных интерферирующих частиц
Дефектные интерферирующие частицы и стандартные вирионы РСВ не удается разделить физическими методами, на проявление устойчивой к ультрафиолетовому облучению и чувствительной к нейтрализующим антителам интерферирующей активности при пассировании вируса с высокой множественностью инфекции свидетельствует о накоплении ДИЧ. Разра-Зотзн колориметрический метод количественного определения ДИЧ в препаратах РСВ. Он основан на измерении интенсивности окрашивания, обусловленного поглощением нейтрального красного клетками, которые выживают при заражении стандартным вирусом в результате защиты ДИЧ. Приводятся данные о том, что колориметрический метод чувствительнее, чем метод, основанный на определении снижения выхода вируса. Колориметрический анализ проводят следующим образом.
1. Монослойные культуры клеток Нер-2, выращенные в лунках планшета диаметром 16 мм, инкубируют с содержащим ДИЧ материалом 2 ч при 37 °С.
2. Содержащий ДИЧ иннокулят удаляют и заражают клетки стандартным вирусом, полученным в результате пассирования с низкой множественностью, и продолжают адсорбцию 2 ч при 37 °С.
3. В каждую лунку вносят по 1 мл культуральной среды и инкубируют клетки 72 ч при 37 °С.
4. Среду удаляют и вносят в каждую лунку по 0,5 мл нейтрального красного в растворе Эрла; инкубацию клеток продолжают еще 2 ч при 37 °С.
5. Краситель удаляют и клетки дважды промывают PBS.
6. Краситель экстрагируют из клеток 1 мл 50% -ного этанола, содержащим 0,1 М NaH2P04, и определяют его количество спектрофотометрически при 540 нм. Полученные данные сравнивают с количеством красителя, экстрагированного из незараженных клеток и из клеток, зараженных стандартным вирусом.
Концентрацию ДИЧ можно рассчитать следующим образом, предполагая, что их распределение по клеткам подчиняется закону Пуассона:
число ДИЧ/мл= 1/разведение, обеспечивающее выживание 63% клеток Х общее число клеток X 1/объем иннокулята.
3.3 Электронная микроскопияПневмовирусы исключительно многообразны по форме; кроме того, их вирионы и нуклеокапсиды нестабильны и легко повреждаются при обычных процедурах концентрирования вирусов. Поэтому точный подсчет вирусных частиц практически невозможен.
Трансмиссионная электронная микроскопия удобна для идентификации вирусов и изучения морфологии и морфогенеза ви-рионов. Сканирующая электронная микроскопия используется для изучения морфологии поверхности зараженных клеток, а также для количественного определения поверхностных антигенов на основе специфического связывания бактериальных клеток.
3.3.1 Морфология вирионовДля негативного контрастирования успешно применяют следующие вещества: фосфорно-вольфрамовая кислота, фосфовольфрамат натрия, фосфовольфрамат калия, кремневольфрамат натрия и уранилацетат. Концентрирование вируса необходимо проводить самым мягким методом, например осаждением полиэтиленгликолем или ультрафильтрацией через фильтры "Амикон", избегая высокоскоростного центрифугирования. Каплю концентрированного раствора вируса наносят на покрытую графитом формваровую сеточку. Для фиксации добавляют равный объем 3% -ного глутарового альдегида в PBS, перемешивают и инкубируют 5 мин при комнатной температуре. Сеточки промывают PBS и проводят негативное контрастирование. Для визуализации нуклеокапсидов зараженные клетки лизируют дистиллированной водой или гипотоническим буферным раствором. Клеточный дебрис можно осадить непосредственно на сеточке перед негативным контрастированием.
3.3.2 Морфогенез вирионов1. Зараженные клетки фиксируют, добавляя к монослою, выращенному в чашке Петри диаметром 50 мм, глутаровый. альдегид до концентрации 2,5%.
2. После фиксации в течение 30 мин монослой дважды промывают PBS и добавляют 10 капель 1% -ного раствора четы-рехокиси осмия.
3. Через 15 мин монослой еще 2 раза промывают PBS, снимают клетки со стекла и суспендируют их в 2 мл PBS.
4. Клетки осаждают центрифугированием и дегидратируют, последовательно суспендируя в растворах с возрастающей концентрацией этанола; в каждом растворе клетки инкубируют 5 мин; при необходимости их можно выдержать в течение ночи в 70% -ном этаноле. После инкубации в 90% -ном этаноле клетки дважды по 15 мин обрабатывают 100% -ным этанолом.
5. Этанол заменяют на смесь этанола и окиси пропилена.
6. Через 15 мин смесь этанол-окись пропилена заменяют на свежую, инкубируют суспензию еще 30 мин, после этого ресуспендируют клетки в смеси этанол-окись пропилена-среда EPON, а затем в среде EPON 1 ч.
7. Компактный осадок клеток наносят на небольшой объем среды EPON в капсуле ВЕЕМ и, заполнив капсулу средой EPON, инкубируют их 48 ч при 60 °С для полимеризации. После этого заключенные в капсулу клетки готовы для приготовления срезов стандартным способом на ультрамикротоме.
8. Тонкие срезы располагают на необработанных сеточках для электронной микроскопии и через 24 ч окрашивают 5 мин,, помещая сеточки препаратами вниз в каплю насыщенного раствора уранилацетата в метаноле.
9. Сеточки промывают дистиллированной водой и сушат на листе фильтровальной бумаги.
10. На срез наносят каплю 0,1 М раствора NaOH и помещают сеточку срезом вниз на 5 мин в каплю раствора ацетата свинца. Сеточки промывают и сушат на листе фильтровальной бумаги, после этого они готовы к электронно-микроскопическому исследованию.
3.3.3 Сканирующая электронная микроскопияДля подготовки клеток к сканирующей электронной микроскопии применяют следующий метод.
1. Клетки, образующие неплотный монослой на покровных стеклах, фиксируют 1-2 ч 2,5% -ным раствором глутарового альдегида в фосфатном буфере.
2. Стекла помещают на 1 ч в 1% -ный раствор четырехокиси осмия в фосфатном буфере.
3. Клетки дегидратируют, последовательно помещая стекла в 30, 50, 70, 90 и 100% -ный этанол, а затем на 15 мин в смесь этанола с ацетоном.
4. Стекла дважды обрабатывают 100% -ным ацетоном и сушат С02 в критической точке, используя аппарат для сушки фирмы Polaran Equipment Ltd., Watford, Herts или другой аналогичный прибор.
5. Покровные стекла фиксируют на алюминиевых подложках с помощью клея "Electrocday 915" и покрывают золотом с помощью диодного напылителя Polaron Е5000 или другой аналогичной системы. Подготовленные таким образом препараты можно исследовать методом сканирующей электронной микроскопии.
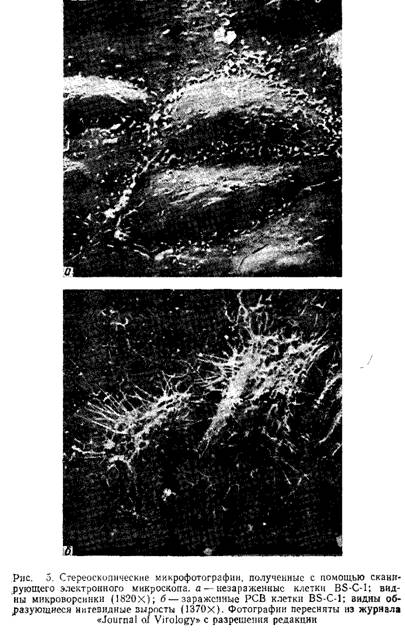
На рис.5 представлена фотография зараженных пневмо-вирусом клеток, выполненная с помощью сканирующей электронной микроскопии. Обратите внимание на длинные выросты инфицированных клеток.
3.3.4 Локализация и количественное определение вирусных антигенов по связыванию стафилококковКлетки Staphylococcus aureus с помощью поверхностного А-белка связываются с Fc-фрагментами иммуноглобулинов. Этот эффект можно использовать для исследования пространственной локализации вирусного антигена на плазматической мембране зараженных клеток после их обработки противовирусной сывороткой. Стафилококки более удобны, чем покрытые А-белком шарики латекса, полиметил-метакрилата или полистирола, в связи с легкостью получения бактерий и одинаковым диаметром всех бактериальных клеток (около 1 мкм). На поверхности клеток, зараженных многими вирусами, стафилококки распределяются равномерно, но в случае пневмовирусов они специфически связываются с нитевидными выростами, образование которых является уникальной особенностью зараженных этими вирусами клеток. Связывание стафилококков высокоспецифично и подтверждает результаты иммунофлуоресцентного окрашивания, которые свидетельствуют о том, что вирусные антигены на поверхности зараженных клеток локализованы преимущественно в составе нитевидных структур.
Связывание стафилококков с поверхностью зараженных клеток проводят по следующей методике.
1. Клетки выращивают на покровных стеклах до образования монослоя и заражают вирусом. Лучше всего использовать клетки почки кенгуровой крысы, поскольку они высокочувствительны к пневмовирусам и не имеют поверхностных выростов.
2. Зараженные клетки инкубируют в поддерживающей среде, промывают PBS для полного удаления входящей в состав среды сыворотки, затем фиксируют 10-15 мин 2,5% -ным раствором глутарового альдегида в PBS при комнатной температуре.
3. Клетки обрабатывают противовирусной сывороткой 1 ч при 37 °С.
4. Антисыворотку удаляют, тщательно промывая клетки PBS. К клеткам добавляют суспензию aureus, штамм Cowan, приготовленную по методу Прингла и Парри, или коммерческий препарат стафилококков и инкубируют 5 мин при 37 °С.
5. Несвязавшиеся стафилококки удаляют, быстро промывая стекла PBS, и клетки фиксируют 60 мин при комнатной температуре 2% -ным раствором четырехокиси осмия в PBS. После этого стекла можно подготовить к сканирующей электронной микроскопии и исследовать.
Подсчет числа связавшихся с клетками стафилококков дает возможность количественно исследовать экспрессию вирусных антигенов на клеточной мембране при условии постановки соответствующих контролей.
3.4 Концентрирование вирусаПредложен простой метод концентрирования РСВ преципитацией полиэтиленгликолем.
1. Готовят 36% -ный раствор ПЭГ 6000 в дистиллированной воде.
2. Раствор охлаждают и добавляют к содержащей вирус жидкости до конечной концентрации ПЭГ 6%.
3. Суспензию инкубируют 2 ч при 4°С.
4. Преципитат собирают центрифугированием при 4°С.
5. Супернатант сливают, осадок дважды промывают буферным раствором и ресуспендируют в небольшом объеме.
3.5 Очистка и радиоактивное мечение РСВКлинические изоляты РСВ в основном ассоциированы с клетками, однако при культивировании некоторых лабораторных штаммов в среду выделяется достаточное количество вируса для успешной очистки. Наиболее удобно использовать для этого штаммы Лонг и А2.
3.5.1 Градиентное центрифугированиеВ литературе можно встретить не одну методику очистки РСВ градиентным центрифугированием. Представленная ниже была успешно использована для очистки штамма А2.
1. Монослой клеток Нер-2 заражают РСВ. Адсорбцию проводят 2 ч при 37°С, затем добавляют культуральную среду.
2. Через 10 ч после заражения добавляют актиномицин D до концентрации 0,3 мкг/мл. Для радиоактивного мечения РНК через 16-20 ч после заражения заменяют культуральную среду на свежую, содержащую 20 мкКи/мл - уридина. Для введения метки в белки в среду добавляют 5 мкКи/мл - метионина.
3. При проявлении ЦПД и образовании синцитиев собирают культуральную жидкость. Клеточный дебрис удаляют центрифугированием 20 мин при 11000 g и концентрируют вирус центрифугированием 90 мин при 65000 g на холоду.
4. Осадок ресуспендируют в буфере NTE при ультразвуковой обработке.
5. Вирус центрифугируют 1 ч при 150 000 g в 10-50% -ном градиенте концентрации сахарозы и собирают фракции, содержащие видимую зону вируса.
6. Содержащие вирус фракции объединяют, разводят NTE и обрабатывают ультразвуком. Вирус вновь центрифугируют 2 ч при 150 000 g в 20-60% -ном градиенте концентрации сахарозы.
7. Вирус концентрируют повторным центрифугированием 90 мин при 65000 g.
Данная методика позволяет получить частично очищенные препараты радиоактивно меченного РСВ. Для последующей очистки предложены более сложные методы; инфекционность, которая в обычных условиях очень чувствительна к центрифугированию, можно стабилизировать добавлением MgS04 до концентрации 1 М.
3.5.2 Очистка РСВ методом изопикнического центрифугирования
Данный метод требует более длительного центрифугирования в градиенте концентрации сахарозы или метризамида. Растворы метризамида обладают более низкой вязкостью и осмотическим давлением, чем растворы сахарозы, и поэтому их использование для изопикнического центрифугирования предпочтительно. Растворы метризамида всегда следует готовить непосредственно перед использованием на 10 мМ HEPES, рН 7,8.
3.6 Радиоактивное мечение вирионной РНКИнтактную вирионную РНК РСВ получить трудно; рекомендуется следующая методика.
Очищенный радиоактивно меченный вирус получают, РНК экстрагируют фенолом и далее из водной фазы осаждают этанолом.
Выделенную РНК очищают центрифугированием в 15 - 30% -ном градиенте концентрации сахарозы в присутствии ДДС-Na в течение 15 ч при 19 000 об/мин.
Альтернативный подход состоит в том, что РНК, экстрагированную из немеченых вирионов, метят по З'-концу с помощью РНК-лигазы и - цитидин-3/,5'-бисфосфата, как описано Вертц и Дэвисом. РНК можно пометить и по 5'-концу после удаления 5'-концевого фосфата щелочной фосфатазой из кишечника крупного рогатого скота, которую затем инактивируют протеиназой К. После этого РНК экстрагируют фенолом, осаждают этанолом и метят в присутствии - ATP и полинуклеотидкиназы из Е. coli, зараженной бактериофагом Т4. Меченую РНК затем отделяют от непрореагировавшего АТР хроматографией на колонке с сефадексом G50. Методика, удобная для мечения РНК РСВ, описана Шубертом и др. .
3.7 Анализ вирусных РНК и белков методом электрофореза в полиакриламидном геле 3.7.1 Анализ белков
Синтезированные in vivo или in vitro меченые белки можно анализировать с помощью электрофореза в 10% -ном или б - 15% -ном градиентном полиакриламидном геле, как описано Марсденом и др. .
После электрофоретического разделения в полиакриламидном геле белки можно перенести на нитроцеллюлозный фильтр для биохимического или иммунологического анализа. Применение этого метода к белкам РСВ описано Хиерхольцером и др. Перенос белков из. полиакриламидного геля на нитроцеллюлозу осуществляется электрофоретически по нижеследующей методике.
1. После электрофоретического разделения белков полиак-риламидный гель помещают в камеру прибора для электрофоретического переноса вместе с листом нитроцеллюлозы. В камеру заливают буферный раствор, содержащий 0,025 М трис, 0, 193 М глицин и 20% метанола, рН 8,35.
2. Для переноса проводят электрофорез при 60 В по крайней мере 4 ч при 4°С.
3. Лист нитроцеллюлозы разрезают на полоски и помещают каждую в закрытую пробирку.
4. Полоски инкубируют с анти-РСВ сывороткой 2 ч при комнатной температуре.
5. Полоски 3 раза промывают PBS, содержащим твин 80.
6. Полоски инкубируют с конъюгатом, разведенным в 1000 раз, 3 ч при комнатной температуре.
7. Полоски 3 раза промывают тем же раствором.
8. Нитроцеллюлозу проявляют раствором, содержащим 50 мг 3,3'-диаминобензидина и 100 мкл 3% -ной перекиси водорода в 100 мл PBS, рН 7,2.
9. Нитроцеллюлозу промывают водой и сушат на листе фильтровальной бумаги.
10. Количественный анализ можно провести спектрофотометрически после обработки нитроцеллюлозы веществами, растворяющими пластмассы. В результате подобной обработки нитроцеллюлоза становится прозрачной.
3.7.2 Анализ РНКРадиоактивно меченную РНК из очищенных вирионов или из цитоплазмы зараженных РСВ клеток можно проанализировать электрофорезом в 1,5% -ном геле агарозы, содержащем 6 М мочевину, по следующей методике.
1. Для приготовления геля размером 50X17X0,3 см готовят отдельно следующие растворы: а) 120 мл 4,5% -ной агарозы; для растворения агарозы суспензию обрабатывают в микроволновой печи или автоклавируют ее; б) 1,5-кратный концентрат раствора мочевины в цитратном буфере; для этого к 129,6 г мочевины добавляют 9 мл концентрированного 1 М цитратного буфера и растворяют, доводя объем водой до 240 мл.
2.1,5-кратный буфер нагревают и смешивают с 3-кратным концентратом агарозы.
3. Гель заливают на холоду в горизонтальный аппарат для электрофореза в 3 стадии, перерывы между стадиями составляют 45 мин.
4. В гель немедленно вводят гребенку.
5. Гель используют для электрофореза через 2-3 ч после заливки. Гребенку вынимают, промывают ячейки буфером с мочевиной и заполняют их тем же буфером. В ячейки вносят образцы, смешанные с буфером для нанесения.
6. Электрофорез проводят на холоду при градиенте напряжения 5 В/см в течение 18 ч. Основные растворы готовят следующим образом.
1 М цитратный буфер, рН 3,0: смешивают 1 М растворы лимонной кислоты и цитрата натрия до достижения рН 3,0. Буфер для электрофореза: 1 М. цитратный буфер разводят в 40 раз, получая 0,025 М цитратный буфер, рН 3,0 Буфер для нанесения образцов: 0,025 мл 1 М цитратного буфера, 3,6 г 6 М мочевины, 2 г 20% -ной сахарозы, 0,1 мл 0,005% -ного бромфенолового синего, объем доводят водой до 10 мл. Буфер с 6 М мочевиной: 3,6 г мочевины, 0,25 мл 1 М цитрата, рН 3,0; объем доводят до 10 мл дистиллированной водой.
3.8 Очистка матричной РНК для трансляцииМатричную РНК для трансляции in vitro получают следующим образом.
1. 50-Ю6 клеток заражают РСВ. Клетки инкубируют 10-12 ч при 37 °С.
2. Монослой промывают, снимают клетки со стекла и суспендируют в буферном растворе. Суспензию оставляют на 10 мин при 4°С для набухания клеток.
3. Клетки разрушают в гомогенизаторе Даунса, затем осаждают клеточный и дебрис центрифугированием при 4°С.;
4. Осадок ресуспендируют в 2 мл того же буфера, вновь гомогенизируют и центрифугируют. Супернатанты, полученные после первого и второго центрифугирований, объединяют.
5. К супернатанту добавляют CsCl и N-лаурилсаркозин. Раствор тщательно перемешивают и прогревают 1-2 мин при 51 °С.
6. 7 мл полученного раствора наслаивают на 2 мл раствора в пробирке для ротора SW40 и добавляют сверху буфер, чтобы заполнить пробирку. Пробирки центрифугируют в роторе SW40 16 ч при 25000 об/мин и 25 °С.
7. Осадок ресуспендируют в 0,5 мл стерильной дистиллированной воды. РНК дважды переосаждают этанолом, добавляя 1,2 мл этанола, 25 мкл 4 М раствора NaCl и 10 мкл 10% -ного ДДС-Na.
8. После второго осаждения осадок ресуспендируют в буфере, добавляют ДДС-Na до концентрации 0,2% и проводят хроматографию на колонке с олиго - целлюлозой. Связавшийся материал элюируют и осаждают РНК этанолом, добавляя в ка* честве носителя тРНК из печени кролика. Осадок ресуспендируют в том же буфере без ДДС-Na и переосаждают РНК двумя объемами этанола в присутствии 0,2 М ацетата калия. Конечный осадок ресуспендируют в 250-500 мкл 0,01 М HEPES, рН 7,6. В качестве бесклеточной системы для синтеза белка можно использовать зависимый от мРНК лизат ретикулоцитов кролика, приготовленный, как описано Престоном, или коммерческий препарат лизата. Для контроля следует параллельно получить мРНК из незараженных клеток. Индивидуальные вирусные мРНК для трансляции в бесклеточной системе можно выделить методом гибридизационной селекции с использованием клонов кДНК с известной структурой, как описано Коллинзом и Вертц.
3.9 Молекулярное клонирование и олигонуклеотидное секвенирование10 генов РСВ были идентифицированы в результате клонирования кДНК с последующим картированием и трансляцией вирусных мРНК - кДНК получали обратной транскрипцией мРНК из зараженных РСВ клеток Нер-2 и клонировали их в Е. coli с использованием плазмиды pBR322.
Нуклеотидная последовательность гена белка нуклеопротеи-на РСВ и кодируемая ею аминокислотная последовательность - опубликованы Венкатесаном и Эланго, а последовательности гена и белка М. - Сатаке и Венкатесаном. В этих работах подробно описаны методы олигонуклеотидного секвенирования в применении к РСВ.
3.10 Очистка вирусных белков 3.10.1 Вирусные полипептиды
В настоящее время очищенные вирусные белки можно получать методом аффинной хроматографии с использованием специфических моноклональных антител. Другой подход состоит в экспрессии клонированных генов в прокариотических или в эукариотических клетках. Однако непосредственно для РСВ такие методики пока не описаны.
3.10.2 Очистка капсидного белкаКапсидный белок выделяют из очищенных нуклеокапсидов, которые в свою очередь можно получить либо из вирионов, либо из экстракта зараженных клеток. Ниже приведена методика выделения из клеточных экстрактов, дающая более высокий выход.
50-Ю6 клеток заражают РСВ. Через 36-48 ч после заражения клетки снимают с поверхности флакона раствором, содержащим 10 мМ трис-НО, рН 7,4, 0,18 М NaCl, 0,25 мМ ЭДТА и 0,1 мМ PMSF. Клетки осаждают центрифугированием и ресуспендируют в 1мл лизирующего буфера 20 мин при 4°С. Клетки разрушают в гомогенизаторе Даунса. К гомогенату добавляют NaCl до концентрации 0,1 М и удаляют неразрушенные клетки, ядра и дебрис центрифугированием 2 мин при 600 g. Нуклеокапсиды осаждают центрифугированием 30 мин при 100000 g. Нуклеокапсиды промывают лизирующим буфером, а затем буфером, содержащим 10 мМ трис-HCl, рН 7,4, и 0,1 мМ ЭДТА. Осадок нуклеокапсидов ресуспендируют и получают очищенные нуклеокапсиды изопикническим центрифугированием в 15-55% -ном градиенте концентрации тартрата калия в буфере ТЕ.
3.10.3 Очистка вирионных гликопротеинов
Предложен метод очистки гликопротеинов РСВ, основанный на солюбилизации вирионов неионным детергентом в буферном растворе с низкой ионной силой.
1. Очищенные методом градиентного центрифугирования вирионы осаждают центрифугированием через слой 30% -ного раствора сахарозы в буфере NTE при 50 000 g 60 мин. Осадок ресуспендируют в 0,01 М фосфатном буфере, содержащем 10% тритона Х-100.
2. Суспензию перемешивают 30 мин при комнатной температуре и центрифугируют 60 мин при 200 000 g.
3. Супернатант экстрагируют п-бутанолом.
4. Экстрагированные белки суспендируют в 0,01 М фосфатном буфере с 1% тритона Х-100 и фракционируют в 5-25% -ном линейном градиенте концентрации сахарозы, содержащем 1% тритона Х-100, 18 ч при 100 000 g. Собирают фракции из верхней части градиента и диализуют их против 0,01 М фосфатнога буфера.
5. Тритон Х-100 удаляют экстракцией п-бутанолом.
6. Фракции, содержащие вирусные гликопротеины, идентифицируют с помощью электрофореза в полиакриламидном геле.
4. Специальные биологические методы 4.1 Получение моноклональных антител
Моноклональные антитела к нескольким белкам РСВ были получены стандартными методами. Ниже приведен типичный протокол.
1. Мышам линии Balb/c внутрибрюшинно вводят зараженные РСВ клетки. Через 10 сут. инокуляцию повторяют.
2. Селезенки иммунизированных мышей суспендируют в среде RPM1 1640, забуференной бикарбонатом натрия и содержащей пенициллин, гентамицин, пируват натрия, L-глютамин и ЭТС.
3. Клетки селезенки иммунизированных мышей сливают с клетками мышиной миеломы, например Balb/c NS1/1, выращиваемой в той же среде. При этом смешивают 3-107 клеток селезенки с таким же количеством клеток миеломы. Клетки центрифугируют 10 мин при 200 g и 1 мин выдерживают при 37 °С. Для слияния клеток по каплям добавляют 1 мл 50% -ного ПЭГ 4000 с 5% ДМСО.
4. Суспензию инкубируют 90 с при 37 °С и останавливают процесс слияния, добавляя 20 мл среды.
5. Клетки осаждают центрифугированием и промывают средой. Осадок ресуспендируют в среде ГАТ и распределяют по плоскодонным лункам планшета для иммунологических реакций.
6. Клетки инкубируют на питательном слое из макрофагов, подготовленном за сутки до слияния; каждая лунка должна содержать 10" перитонеальных макрофагов здоровой мыши.
7. Через 6 сут после начала инкубации в каждую лунку вносят по 50 мкл среды ГАТ.
8. Через 9-12 сут после слияния проверяют активность и специфичность продуцируемых антител методом ELISA с антигенами РСВ.
9. Гибридомы, продуцирующие специфические антитела, клонируют методом предельного разведения в планшете для иммунологических реакций, используя макрофаги или клетки селезенки в качестве фидера. Клонирование проводят дважды. Гибридомы считают стабильными, если 95% клонов продуцируют специфические антитела.
4.2 Выделение температурно-чувствительных мутантовДля анализа функций генов и биохимических исследований полезно иметь температурно-чувствительные мутанты вируса. Их выделяют следующим образом.
1.106 клеток BS-C-1, выращенных в чашке Петри диаметром 50 мм или во флаконе с завинчивающейся крышкой емкостью 30 мл, заражают РСВ дикого типа с множественностью инфекции 0,01 БОЕ/кл. Адсорбцию проводят 1 ч при 31 °С.
2. Вирус отмывают двумя сменами среды и добавляют 3 мл среды Игла, содержащей 2,5% ЭТС и мутаген.
3. Культуру инкубируют при 31 °С до проявления ЦПД в контрольной зараженной культуре, к которой мутаген не добавляли.
4. Разведения культуральной жидкости из обработанных мутагеном культур титруют методом бляшек после осветления, но без замораживания и оттаивания. Для этого на клетки наносят необходимые разведения и инкубируют клетки, под 0,9% -ным агаровым покрытием 5-7 сут при 31 °С.
5. Вирус из полученных индивидуальных бляшек наращивают, перенося бляшки пастеровской пипеткой на свежие монослойные культуры.
6. Проводят скрининг полученных изолятов вируса на способность давать бляшки при 31 и 39 °С. Для этого зараженные чашки инкубируют параллельно при каждой температуре. Альтернативный метод состоит в прямом скрининге бляшек, образованных вирусом, обработанным мутагеном. Для этого 2 чашки с культурой, покрытой слоем 0,9% -ного агара толщиной 2 мм, делят на секторы и в каждый сектор помещают 1 бляшку., После адсорбции в течение 30 мин при 31 °С заливают второй слой агарового покрытия. После этого одну чашку инкубируют при 31 °С, а другую при 39 °С. Из секторов, где бляшки образуются при 31 °С, но не при 39 °С, их извлекают для дополнительной проверки.
Температурно-чувствительные мутанты можно подразделить на функциональные группы с помощью теста комплементации, факторы, влияющие на комплементацию, детально изучены.
Как и для остальных вирусов с негативной полярностью РНК и несегментированным геномом, рекомбинация между температурно-чувствительными мутантами РСВ, принадлежащими к одной или к разным группам комплементации, не описана.
4.3 Получение персистентно инфицированных клеточных культурПерсистентной инфекции культур клеток при полном отсутствии ЦПД или минимальном его проявлении можно добиться несколькими способами. Одним из них является пассирование вируса с высокой множественностью инфекции и пересев выживающих клеток. Другие методы включают заражение частично устойчивых клеток в присутствии противовирусных антител или интерферона. Для получения персистентно инфицированных культур можно инкубировать клетки, зараженные температурно-чувствительными мутантами РСВ при частично ограничивающей размножение температуре для подавления ЦПД. Такие культуры после установления персистенции поддерживают при той же температуре. Исход опыта зависит от конкретных свойств используемого мутанта. Температурно-чувствительные мутанты комплементационной группы В, продуцирующие при неразрешающей температуре лишь небольшое количество антигена, не подходят для данной цели, поскольку дают либо абортивную, либо литическую инфекцию. С другой стороны, мутанты группы D, синтезирующие при неразрешающей температуре значительное количество антигена, воспроизводимо дают персистентно инфицированные культуры, которые можно пассировать неограниченное число раз. Персистентно инфицированные культуры служат хорошим источником вирусного антигена для проведения анализа по методу ELISA.
0 комментариев