Энтальпия образования (enthalpy of formation) является основным свойством, используемым при решении многих теоретических и практических задач. Знание энтальпий образования реагентов позволяет вычислить тепловые эффекты интересующих реакций, что необходимо при оценке адиабатического перепада температур в зоне реакции, формировании требований к конструкции реактора и технологическим особенностям химического процесса. Энтальпии образования веществ необходимы при выполнении количественного термодинамического анализа процессов, определении теоретической степени конверсии реагентов, выборе условий проведения химического превращения и т.п. Качество выполненного термодинамического анализа во многом зависит от надежности сведений по энтальпиям образования веществ.
Единицами измерения энтальпии являются кДж/моль и Дж/моль. В справочной литературе прежних лет энтальпии образования часто представлены в ккал/моль (1 кал = 4,184 Дж, 1 ккал = 4,184 кДж).
Энтальпия образования (![]() ) соединения в узком смысле есть стандартное изменение энтальпии в реакции образования данного вещества из элементов или простых веществ. Простыми веществами являются химические элементы, находящиеся при рассматриваемой температуре в их естественном фазовом и химическом состояниях.
) соединения в узком смысле есть стандартное изменение энтальпии в реакции образования данного вещества из элементов или простых веществ. Простыми веществами являются химические элементы, находящиеся при рассматриваемой температуре в их естественном фазовом и химическом состояниях.
В качестве стандартного состояния вещества выбирается такое его состояние, при котором это вещество устойчиво при стандартном давлении, равном 1 атм (101325 Па). Конденсированное состояние вещества является стандартным вплоть до тех температур, при которых давление его насыщенного пара достигает 1 атм. Выше этих температур в качестве стандартного выбирается состояние идеального газа.
Для простых веществ, участвующих в формировании молекул большинства органических соединений, стандартным состоянием при 298,15 К является:
· для углерода - графит;
· для водорода, кислорода, азота, фтора и хлора - идеальный двухатомный газ;
· для брома - двухатомная жидкость;
· для иода и серы - кристаллическое состояние, двухатомное и одноатомное соответственно.
Абсолютные значения энтальпий не могут быть определены, поскольку они включают абсолютные значения внутренней энергии. Необходимость определения энтальпий образования соединений потребовала достижения международного соглашения, по которому были приняты равными нулю значения ![]() элементов и простых веществ, находящихся в стандартном состоянии.
элементов и простых веществ, находящихся в стандартном состоянии.
Величина и знак ![]() веществ со сложным строением молекул могут быть различными. Объясняется это следующим. Образование вещества из свободных атомов всегда сопровождается выделением энергии,
веществ со сложным строением молекул могут быть различными. Объясняется это следующим. Образование вещества из свободных атомов всегда сопровождается выделением энергии, ![]() полученных при этом веществ отрицательна. Однако при образовании вещества из простых веществ, состоящих из двухатомных молекул (H2, О2, N2, Cl2 и пр.) или находящихся в конденсированном состоянии (углерод, бром и т.п.), некоторое количество энергии требуется для разрыва связей в молекулах этих простых веществ или для перевода их в газообразное состояние. В результате этого энтальпия образования может быть и положительной, и отрицательной, и равной нулю величиной.
полученных при этом веществ отрицательна. Однако при образовании вещества из простых веществ, состоящих из двухатомных молекул (H2, О2, N2, Cl2 и пр.) или находящихся в конденсированном состоянии (углерод, бром и т.п.), некоторое количество энергии требуется для разрыва связей в молекулах этих простых веществ или для перевода их в газообразное состояние. В результате этого энтальпия образования может быть и положительной, и отрицательной, и равной нулю величиной.
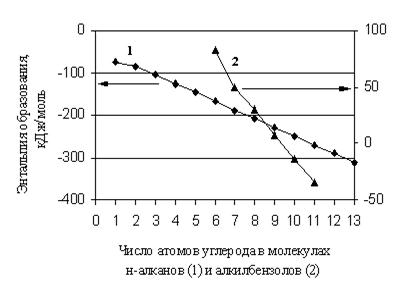
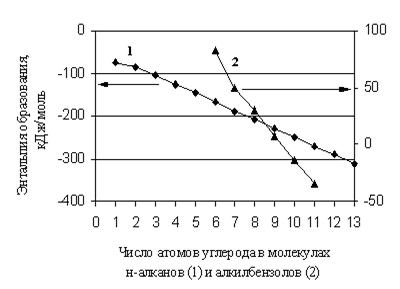
В общем случае значение и знак ![]() не дают оснований для каких-либо утверждений относительно термодинамической стабильности вещества, так как она зависит не только от энтальпийной, но и от энтропийной составляющей изменения свободной энергии при образовании этого вещества. Тем не менее, для сопоставления термодинамической стабильности представителей одной гомологической группы или соединений с близким строением молекул может быть достаточно информативным, в первом приближении, анализ их энтальпий образования. В этом случае вещество, имеющее меньшее значение энтальпии образования, обладает большей термодинамической стабильностью.
не дают оснований для каких-либо утверждений относительно термодинамической стабильности вещества, так как она зависит не только от энтальпийной, но и от энтропийной составляющей изменения свободной энергии при образовании этого вещества. Тем не менее, для сопоставления термодинамической стабильности представителей одной гомологической группы или соединений с близким строением молекул может быть достаточно информативным, в первом приближении, анализ их энтальпий образования. В этом случае вещество, имеющее меньшее значение энтальпии образования, обладает большей термодинамической стабильностью.
Для большинства соединений реакции их образования из простых веществ не могут быть осуществлены на практике. Основным источником фактической информации об энтальпиях образования органических соединений являются экспериментальные данные по энтальпиям их сгорания (![]() ), полученные калориметрическим методом. Накопленные к настоящему времени и рекомендуемые значения
), полученные калориметрическим методом. Накопленные к настоящему времени и рекомендуемые значения ![]() и
и ![]() содержатся в компиляциях [1-4]. До настоящего времени справочные сведения об
содержатся в компиляциях [1-4]. До настоящего времени справочные сведения об ![]() были представлены исключительно калориметрическими данными. Современные эмпирические методы прогнозирования
были представлены исключительно калориметрическими данными. Современные эмпирические методы прогнозирования ![]() также базируются только на калориметрических данных. При этом следует отметить, что информация, уникальная по спектру задействованных веществ, объему выполненных исследований и точности полученных термодинамических характеристик, содержится в результатах изучения химического равновесия. На наш взгляд, эффективное использование этих сведений позволит существенно расширить прогностические возможности методов массовых расчетов
также базируются только на калориметрических данных. При этом следует отметить, что информация, уникальная по спектру задействованных веществ, объему выполненных исследований и точности полученных термодинамических характеристик, содержится в результатах изучения химического равновесия. На наш взгляд, эффективное использование этих сведений позволит существенно расширить прогностические возможности методов массовых расчетов ![]() органических веществ.
органических веществ.
Прогнозирование
Идеология метода изложена в [8-10]. В свое время он был достаточно широко апробирован на различных свойствах в приближении неполного второго окружения атомов. Яровым [11] выполнен анализ возможностей различных приближений метода Татевского и рекомендован набор параметров для прогнозирования основных термодинамических и физико-химических свойств веществ. К настоящему времени накоплен значительный дополнительный объем фактического материала, что позволяет на более широкой основе рассмотреть возможности метода Татевского и, в случае необходимости, уточнить некоторые из его параметров. Используемая при этом процедура подробно изложена для одного класса соединений - алканов, для других классов поясняются только некоторые специфические особенности расчетной схемы, обусловленные строением молекул. Сопоставление результатов производится с методом Бенсона и между различными приближениями метода Татевского.
В рассматриваемом методе в качестве структурной единицы избрана углерод-углеродная связь. При этом парциальные вклады, соответствующие водород-углеродным связям, приняты равными нулю из-за наличия линейных соотношений между числом углерод-углеродных и водород-углеродных связей [11]. Отличительными особенностями метода являются его исключительная простота в использовании и высокая точность прогноза в большинстве случаев.
Алканы
Выбор алканов для иллюстрации подходов к прогнозированию очевиден, поскольку алкильная составляющая с определенным весом входит в состав молекулы большинства органических соединений, и достоверность прогноза свойств интересующих веществ во многом зависит от качества прогноза свойств алканов. Таким образом, при формировании любого аддитивного метода для свойств органических веществ, прежде всего, необходимо установить зависимость свойств алканов от строения их молекул [50-51].
Первый и очень важный этап работы состоит в создании базы данных, которая должна быть предельно полной на настоящий момент и сопровождаться информацией о погрешностях величин и использованных в оригинальных работах экспериментальных и (или) расчетных методах. Как правило, значительная часть этих сведений содержится в различных компиляциях и монографиях, где указано, за какой период обобщен фактический материал, и в некоторых случаях дается его критический анализ. База данных по ![]() алканов наиболее представительна. В табл. 1.9 обобщены сведения для 161 соединения с различным строением молекул, заимствованные из [2, 5, 27, 28].
алканов наиболее представительна. В табл. 1.9 обобщены сведения для 161 соединения с различным строением молекул, заимствованные из [2, 5, 27, 28].
Весь массив разбит на три блока в зависимости от уровня погрешностей приведенных значений. Для линейных алканов погрешность определения ![]() составляет 0,4 – 1,0 кДж/моль [2]. Для разветвленных структур, отмеченных в табл. 1.9 знаком (*), она находится в диапазоне 0,4 – 2,0 кДж/моль [2]. Для разветвленных алканов третьего блока погрешность либо не приведена [27], либо достигает 4,3 кДж/моль [28]. Уровень погрешностей определил тактику использования фактического материала, которая состоит в следующем.
составляет 0,4 – 1,0 кДж/моль [2]. Для разветвленных структур, отмеченных в табл. 1.9 знаком (*), она находится в диапазоне 0,4 – 2,0 кДж/моль [2]. Для разветвленных алканов третьего блока погрешность либо не приведена [27], либо достигает 4,3 кДж/моль [28]. Уровень погрешностей определил тактику использования фактического материала, которая состоит в следующем.
На этапе определения необходимой и достаточной глубины детализации метода рассматривались следующие три уровня описания свойства:
уровень 1 – значимыми являются только взаимодействия валентно-связанных атомов углерода;
уровень 2 – значимыми являются взаимодействия валентно-связанных атомов углерода и атомов углерода, разделенных двумя связями;
уровень 3 – значимыми являются взаимодействия валентно-связанных атомов углерода и атомов углерода, разделенных двумя и тремя связями.
Расчет парциальных вкладов производился с применением пакета программ “Excel” в режиме “Поиск решения” при минимизации ![]() . Для определения значений парциальных вкладов (С1-С1)1, (С1-С2)1 и (С2-С2)1 использованы только
. Для определения значений парциальных вкладов (С1-С1)1, (С1-С2)1 и (С2-С2)1 использованы только ![]() для линейных алканов как обладающие наибольшей достоверностью. Полученные при этом величины, приведенные в табл. 1.10, сохранялись неизменными при определении значений прочих парциальных вкладов для всех уровней детализации метода. Обработке подвергался весь массив экспериментальных данных, для которых известна экспериментальная погрешность определения энтальпий образования и которые отмечены в табл. 1.9 знаком (*). Энтальпия образования каждого вещества при этом была представлена как сумма парциальных вкладов вида (Ci-Cj)1, соответствующих углерод-углеродным связям всех разновидностей. Например,
для линейных алканов как обладающие наибольшей достоверностью. Полученные при этом величины, приведенные в табл. 1.10, сохранялись неизменными при определении значений прочих парциальных вкладов для всех уровней детализации метода. Обработке подвергался весь массив экспериментальных данных, для которых известна экспериментальная погрешность определения энтальпий образования и которые отмечены в табл. 1.9 знаком (*). Энтальпия образования каждого вещества при этом была представлена как сумма парциальных вкладов вида (Ci-Cj)1, соответствующих углерод-углеродным связям всех разновидностей. Например,
![]() (2-Метилбутана)= 2·(C1-C3)1+1·(C1-C2)1+1·(C2-C3)1,
(2-Метилбутана)= 2·(C1-C3)1+1·(C1-C2)1+1·(C2-C3)1,
![]() (3,3,4,4-Тетраметилгексана)= 2·(C1-C2)1+2·(C2-C4)1+4·(C1-C4)1 +
(3,3,4,4-Тетраметилгексана)= 2·(C1-C2)1+2·(C2-C4)1+4·(C1-C4)1 +
+1·(C4-C4)1 и т.п.
Таблица 1.9
Результаты прогнозирования| Соединение | кДж/моль | |||||
| ΔfH0g, 298 (эксп.) | ΔfH0g, 298 (расч.)- ΔfH0g, 298 (эксп.) | |||||
| По Бен-сону | 1 уровень | 2 уровень | 3 уровень | Для напряжен-ных структур | ||
| Этан* | -83,825 | -0,56 | 0,00 | 0,00 | 0,00 | 0,00 |
| Пропан* | -104,685 | -0,34 | -0,48 | -0,48 | -0,48 | -0,48 |
| Бутан* | -125,795 | 0,13 | 0,00 | 0,00 | 0,00 | 0,00 |
| Пентан* | -146,765 | 0,46 | 0,34 | 0,34 | 0,34 | 0,34 |
| Гексан* | -166,925 | -0,02 | -0,13 | -0,13 | -0,13 | -0,13 |
| Гептан* | -187,805 | 0,22 | 0,12 | 0,12 | 0,12 | 0,12 |
| Октан* | -208,755 | 0,53 | 0,45 | 0,45 | 0,45 | 0,45 |
| Нонан* | -228,865 | 0,00 | -0,07 | -0,07 | -0,07 | -0,07 |
| Декан* | -249,535 | 0,03 | -0,03 | -0,03 | -0,03 | -0,03 |
| Ундекан* | -270,165 | 0,02 | -0,03 | -0,03 | -0,03 | -0,03 |
| Додекан* | -290,795 | 0,01 | -0,03 | -0,03 | -0,03 | -0,03 |
| Тридекан* | -311,425 | 0,00 | -0,02 | -0,02 | -0,02 | -0,02 |
| Тетрадекан* | -332,055 | -0,01 | -0,02 | -0,02 | -0,02 | -0,02 |
| Пентадекан* | -352,685 | -0,02 | -0,02 | -0,02 | -0,02 | -0,02 |
| Гексадекан* | -373,315 | -0,03 | -0,02 | -0,02 | -0,02 | -0,02 |
| Гептадекан* | -393,945 | -0,04 | -0,02 | -0,02 | -0,02 | -0,02 |
| Октадекан* | -414,575 | -0,05 | -0,02 | -0,02 | -0,02 | -0,02 |
| Нонадекан* | -435,205 | -0,06 | -0,01 | -0,01 | -0,01 | -0,01 |
| Эйкозан* | -455,835 | -0,07 | -0,01 | -0,01 | -0,01 | -0,01 |
| Среднее абсолютное отклонение |
| 0,14 | 0,10 | 0,10 | 0,10 | 0,10 |
| 2-Метилбутан* | -153,702 | -1,5 | 0,2 | 0,2 | 0,2 | 0,2 |
| 2,2-Диметилпропан* | -168,492 | 1,8 | 3,3 | 0,1 | 0,1 | 0,1 |
| 2-Метилпентан* | -174,555 | 2,1 | 0,4 | 0,4 | 0,4 | 0,4 |
| 3-Метилпентан* | -172,005 | 2,9 | 0,3 | 0,5 | 0,5 | 0,5 |
| 2,3-Диметилбутан* | -175,905 | -2,1 | 0,4 | -0,2 | -0,2 | -0,2 |
| 2,2-Диметилбутан* | -183,975 | 3,4 | 2,4 | -0,3 | -0,3 | -0,3 |
| 2-Метилгексан* | -194,605 | 1,5 | -0,2 | -0,1 | -0,1 | -0,1 |
| 3-Метилгексан* | -191,305 | 1,6 | -1,0 | -0,9 | -0,9 | -0,9 |
| 3-Этилпентан* | -189,505 | 3,1 | -0,3 | -0,1 | -0,1 | -0,1 |
| 2,3-Диметилпентан* | -194,105 | -1,2 | 0,5 | -0,1 | -0,1 | -0,1 |
| 2,4-Диметилпентан* | -201,675 | 3,1 | -0,2 | -0,1 | -0,1 | -0,1 |
| 2,2-Диметилпентан* | -205,805 | 4,6 | 3,6 | 0,9 | 1,6 | 0,9 |
| 3,3-Диметилпентан* | -201,405 | 6,9 | 3,5 | 1,2 | 1,2 | 1,2 |
| 2,2,3-Триметилбутан* | -204,405 | 1,0 | 3,6 | 1,9 | 1,9 | 1,9 |
| 2-Метилгептан* | -215,352 | 1,6 | -0,1 | 0,0 | 0,0 | 0,0 |
| 3-Метилгептан* | -212,505 | 2,1 | -0,4 | -0,3 | -0,3 | -0,3 |
| 4-Метилгептан* | -211,965 | 1,6 | 1,0 | -0,8 | -0,8 | -0,8 |
| 3-Этилгексан* | -210,715 | 3,7 | 0,3 | 0,5 | 0,5 | 0,5 |
| 2,3-Диметилгексан* | -213,805 | -2,1 | -0,4 | -1,0 | -1,0 | -1,0 |
| 2,4-Диметилгексан* | -219,245 | 3,4 | -0,8 | -0,6 | -0,6 | -0,6 |
Продолжение табл. 1.9
| Соединение | кДж/моль | |||||
| ΔfH0g, 298 (эксп.) | ΔfH0g, 298 (расч.)- ΔfH0g, 298 (эксп.) | |||||
| По Бен-сону | 1 уровень | 2 уровень | 3 уровень | Для напряжен-ных структур | ||
| 2,5-Диметилгексан* | -222,515 | 3,3 | 0,0 | 0,1 | 0,1 | 0,1 |
| 3,4-Диметилгексан* | -212,675 | 0,1 | 0,9 | 0,4 | 0,4 | 0,4 |
| 3-Этил-2-метилпентан* | -212,805 | 0,3 | 1,0 | 0,5 | 0,5 | 0,5 |
| 2,3,4-триметилпентан* | -217,325 | -4,1 | 1,7 | 0,4 | 0,4 | 0,4 |
| 2,2-Диметилгексан* | -224,605 | 2,7 | 1,8 | -0,9 | -0,7 | -0,9 |
| 3,3-Диметилгексан* | -219,995 | 4,8 | 1,5 | -0,8 | -0,1 | -0,8 |
| 3-Метил-3-этилпентан* | -214,855 | 6,4 | 0,6 | -1,2 | -1,2 | -1,2 |
| 2,2,3-Триметилпентан* | -219,955 | -0,7 | 1,1 | -0,7 | 0,1 | -0,7 |
| 2,2,4-Триметилпентан* | -224,015 | -3,4 | -5,9 | -0,7 | -0,5 | -1,2 |
| 2,3,3-Триметилпентан* | -218,455 | 1,1 | 1,3 | 0,0 | 0,0 | 0,0 |
| 2,2,3,3-Тетраметилбутан* | -225,222 | -3,6 | 2,9 | 2,6 | 2,6 | -4,4 |
| 2-Метилоктан* | -235,855 | 1,5 | -0,2 | -0,1 | -0,1 | -0,1 |
| 2,2-Диметилгептан* | -246,105 | 3,6 | 2,7 | -0,1 | 0,1 | -0,1 |
| 2,2,5-Триметилгексан* | -253,265 | 5,2 | 2,7 | 0,0 | -0,4 | 0,0 |
| 3,3-Диэтилпентан* | -231,842 | 9,4 | 1,2 | -0,1 | -0,1 | -0,1 |
| 2,2,3,3-Тетраметилпентан* | -237,112 | -5,7 | -1,6 | -1,4 | -0,7 | 1,3 |
| 2,2,3,4-Тетраметилпентан* | -234,972 | -11,9 | -5,9 | -0,4 | -0,3 | -0,2 |
| 2,2,4,4-Тетраметилпентан* | -242,252 | -14,0 | -15,6 | 0,0 | 0,0 | -0,6 |
| 2,3,3,4-Тетраметилпентан* | -236,312 | -3,8 | -0,1 | -0,4 | -0,4 | -0,4 |
| 3,3,5-Триметилгептан* | -259,872 | 1,2 | -4,5 | 1,2 | 0,8 | 0,7 |
| 2,2,5-Триметилгептан* | -272,212 | 6,9 | 3,6 | 0,9 | 0,4 | 0,9 |
| 2,2,3,3-Тетраметилгексан* | -257,992 | -5,4 | -1,3 | -1,2 | -0,2 | 1,5 |
| 2,2,5,5-Тетраметилгексан* | -285,892 | 9,1 | 7,4 | 1,9 | 0,0 | 1,9 |
| Среднее абсолютное отклонение* |
| 3,7 | 2,0 | 0,6 | 0,5 | 0,7 |
| 3-Метилоктан | -233,727 | 2,7 | 0,2 | 0,3 | 0,3 | 0,3 |
| 4-Метилоктан | -235,227 | 4,2 | 1,7 | 1,8 | 1,8 | 1,8 |
| 2,3-Диметилгептан | -235,927 | -0,6 | 1,0 | 0,4 | 0,4 | 0,4 |
| 2,4-Диметилгептан | -241,027 | 4,5 | 0,3 | 0,5 | 0,5 | 0,5 |
| 2,5-Диметилгептан | -240,727 | 4,2 | 0,0 | 0,2 | 0,2 | 0,2 |
| 2,6-Диметилгептан | -242,827 | 2,9 | -0,4 | -0,3 | -0,3 | -0,3 |
| 3,4-Диметилгептан | -233,827 | 0,6 | 1,4 | 0,9 | 0,9 | 0,9 |
| 3,5-Диметилгептан | -238,927 | 5,7 | 0,7 | 1,0 | 1,0 | 1,0 |
| 3,3-Диметилгептан | -241,527 | 5,7 | 2,3 | 0,1 | 0,3 | 0,1 |
| 4,4-Диметилгептан | -241,227 | 5,4 | 2,0 | -0,2 | 1,3 | -0,2 |
| 3-Этилгептан | -231,527 | 3,8 | 0,4 | 0,6 | 0,6 | 0,6 |
| 4-Этилгептан | -231,827 | 4,1 | 0,7 | 0,9 | 0,9 | 0,9 |
| 2,3,4-Триметилгексан | -235,427 | -3,3 | 1,7 | 0,5 | 0,5 | 0,5 |
| 2,3,5-Триметилгексан | -242,627 | 0,6 | 0,6 | 0,1 | 0,1 | 0,1 |
| 2,4,4-Триметилгексан | -240,027 | -1,3 | -6,3 | -0,6 | -0,4 | -1,1 |
| 3,3,4-Триметилгексан | -235,427 | 0,8 | 0,1 | -1,1 | -0,3 | -1,1 |
| 3-Этил-2-метилгексан | -234,627 | 1,4 | 2,2 | 1,7 | 1,7 | 1,7 |
| 3-Этил-3-метилгексан | -231,327 | 2,2 | -3,6 | -5,4 | -4,6 | -5,4 |
Продолжение табл. 1.9
| Соединение | кДж/моль | |||||
| ΔfH0g, 298 (эксп.) | ΔfH0g, 298 (расч.)- ΔfH0g, 298 (эксп.) | |||||
| По Бен-сону | 1 уровень | 2 уровень | 3 уровень | Для напряжен-ных структур | ||
| 3-Этил-4-метилгексан | -238,727 | 8,9 | 8,8 | 8,4 | 8,4 | 8,4 |
| 4-Этил-2-метилгексан | -236,227 | 3,0 | -2,0 | -1,7 | -1,7 | -1,7 |
| 3-Этил-2,2-диметилпентан | -231,327 | -6,7 | -5,7 | -7,4 | -5,9 | -7,4 |
| 3-Этил-2,3-диметилпентан | -233,827 | 2,5 | 0,3 | -0,5 | -0,5 | -0,5 |
| 3-Этил-2,4-диметилпентан | -227,927 | -10,8 | -5,8 | -7,0 | -7,0 | -7,0 |
| 2,2,3-Триметилгексан | -241,427 | 0,1 | 1,9 | 0,2 | 0,4 | 0,2 |
| 2,3,3-Триметилгексан | -239,327 | -2,0 | 1,5 | 0,2 | 1,0 | 0,2 |
| 2,2,4-Триметилгексан | -243,227 | -1,5 | -4,8 | 0,4 | 0,0 | -0,1 |
| 2-Метилнонан | -256,527 | 1,5 | -0,2 | -0,1 | -0,1 | -0,1 |
| 3- Метилнонан | -254,427 | 2,7 | 0,2 | 0,3 | 0,3 | 0,3 |
| 4- Метилнонан | -254,727 | 3,0 | 0,5 | 0,6 | 0,6 | 0,6 |
| 5- Метилнонан | -254,727 | 3,0 | 0,5 | 0,6 | 0,6 | 0,6 |
| 3,3-Диэтилгексан | -252,327 | 9,2 | 1,0 | -0,3 | 0,5 | -0,3 |
| 3,4-Диэтилгексан | -249,727 | 2,6 | 1,7 | 1,3 | 1,3 | 1,3 |
| 3,3-Диэтил-2-метилпентан | -249,727 | 4,5 | -0,2 | -0,5 | -0,5 | -0,5 |
| 2,4-Диметил-3-(1-метилэтил)-пентан | -238,927 | -25,9 | -16,8 | -18,7 | -18,7 | -18,7 |
| 2,2-Диметилоктан | -267,027 | 3,8 | 3,0 | 0,2 | 0,4 | 0,2 |
| 2,3-Диметилоктан | -256,627 | -0,6 | 1,1 | 0,5 | 0,5 | 0,5 |
| 2,4-Диметилоктан | -262,027 | 4,8 | 0,7 | 0,9 | 0,9 | 0,9 |
| 2,5-Диметилоктан | -261,727 | 4,5 | 0,4 | 0,6 | 0,6 | 0,6 |
| 2,6-Диметилоктан | -261,427 | 4,2 | 0,1 | 0,3 | 0,3 | 0,3 |
| 2,7-Диметилоктан | -263,527 | 3,0 | -0,3 | -0,2 | -0,2 | -0,2 |
| 3,3-Диметилоктан | -262,227 | 5,7 | 2,4 | 0,1 | 0,3 | 0,1 |
| 3,4-Диметилоктан | -254,527 | 0,7 | 1,5 | 1,0 | 1,0 | 1,0 |
| 3,5-Диметилоктан | -259,927 | 6,1 | 1,1 | 1,3 | 1,3 | 1,3 |
| 3,6-Диметилоктан | -259,227 | 5,4 | 0,4 | 0,6 | 0,6 | 0,6 |
| 4,4-Диметилоктан | -262,227 | 5,7 | 2,4 | 0,1 | 1,1 | 0,1 |
| 4,5-Диметилоктан | -254,827 | 1,0 | 1,8 | 1,3 | 1,3 | 1,3 |
| 3-Этил-2,2-диметилгексан | -252,527 | -6,1 | -5,2 | -6,8 | -5,3 | -6,8 |
| 3-Этил-2,3-диметилгексан | -254,627 | -4,0 | 0,5 | -0,4 | 0,4 | -0,4 |
| 3-Этил-2,4-диметилгексан | -246,327 | -13,0 | -5,6 | -6,7 | -6,7 | -6,7 |
| 3-Этил-2,5-диметилгексан | -261,627 | 2,3 | 1,5 | 1,0 | 1,0 | 1,0 |
| 3-Этил-3,4-диметилгексан | -251,827 | 3,2 | 0,1 | -0,6 | 0,2 | -0,6 |
| 4-Этил-2,2-диметилгексан | -262,427 | 0,4 | -3,8 | 1,5 | 0,6 | 1,0 |
| 4-Этил-3,3-диметилгексан | -246,627 | -5,3 | -6,8 | -8,0 | -6,5 | -8,0 |
| 4-Этил-2,3-диметилгексан | -253,927 | -2,1 | 2,1 | 0,9 | 0,9 | 0,9 |
| 4-Этил-2,4-диметилгексан | -256,427 | 1,1 | -6,3 | -0,1 | 0,1 | -0,6 |
| 3-Этил-2-метилгептан | -254,427 | 0,6 | 1,4 | 0,9 | 0,9 | 0,9 |
| 3-Этил-3-метилгептан | -257,227 | 7,4 | 1,7 | -0,1 | 0,6 | -0,1 |
| 3-Этил-4-метилгептан | -252,327 | 1,8 | 1,8 | 1,3 | 1,3 | 1,3 |
| 3-Этил-5-метилгептан | -257,327 | 6,8 | 1,0 | 1,3 | 1,3 | 1,3 |
| 4-Этил-2-метилгептан | -257,327 | 3,5 | -1,5 | -1,3 | -1,3 | -1,3 |
Продолжение табл. 1.9
| Соединение | кДж/моль | |||||
| ΔfH0g, 298 (эксп.) | ΔfH0g, 298 (расч.)- ΔfH0g, 298 (эксп.) | |||||
| По Бен-сону | 1 уровень | 2 уровень | 3 уровень | Для напряжен-ных структур | ||
| 4-Этил-3-метилгептан | -252,327 | 1,8 | 1,8 | 1,3 | 1,3 | 1,3 |
| 4-Этил-4-метилгептан | -252,327 | 2,5 | -3,3 | -5,0 | -3,5 | -5,0 |
| 5-Этил-2-метилгептан | -256,927 | 3,1 | -1,9 | -1,7 | -1,7 | -1,7 |
| 3-Этилоктан | -252,127 | 3,8 | 0,4 | 0,6 | 0,6 | 0,6 |
| 4-Этилоктан | -252,427 | 4,1 | 0,7 | 0,9 | 0,9 | 0,9 |
| 3-Этил-2,2,3-триметилпентан | -239,727 | -17,0 | -15,4 | -14,7 | -13,2 | -2,3 |
| 3-Этил-2,2,4-триметилпентан | -241,527 | -22,6 | -17,5 | -12,0 | -11,0 | -11,7 |
| 3-Этил-2,3,4-триметилпентан | -243,627 | -7,1 | -9,2 | -9,0 | -9,0 | -9,0 |
| 4-(1-Метилэтил)-гептан | -254,627 | 0,8 | 1,6 | 1,1 | 1,1 | 1,1 |
| 2-Метил-3-(1-Метилэтил)-гексан | -248,727 | -10,6 | -5,6 | -6,9 | -6,9 | -6,9 |
| 2,2,3,3,4-Пентаметилпентан | -241,327 | -24,3 | -16,6 | -7,6 | -7,5 | 0,0 |
| 2,2,3,4,4-Пентаметилпентан | -247,727 | -24,6 | -18,4 | -0,8 | -0,8 | 10,7 |
| 4-Пропилгептан | -252,827 | 4,5 | 1,1 | 1,3 | 1,3 | 1,3 |
| 2,2,3-Триметилгептан | -262,927 | 0,9 | 2,8 | 1,0 | 1,2 | 1,0 |
| 2,2,4-Триметилгептан | -264,927 | -0,4 | -3,8 | 1,5 | 1,1 | 1,0 |
| 2,2,6-Триметилгептан | -274,027 | 5,3 | 2,9 | 0,1 | 0,4 | 0,1 |
| 2,3,3-Триметилгептан | -260,227 | 1,6 | 1,8 | 0,5 | 0,7 | 0,5 |
| 2,3,4-Триметилгептан | -256,427 | -2,9 | 2,1 | 0,8 | 0,8 | 0,8 |
| 2,3,5-Триметилгептан | -261,827 | 2,5 | 1,7 | 1,2 | 1,2 | 1,2 |
| 2,3,6-Триметилгептан | -263,627 | 0,9 | 1,0 | 0,5 | 0,5 | 0,5 |
| 2,4,4-Триметилгептан | -261,327 | -0,7 | -5,6 | 0,0 | 1,0 | -0,5 |
| 2,4,5-Триметилгептан | -261,827 | -0,9 | 1,7 | 1,2 | 1,2 | 1,2 |
| 2,4,6-Триметилгептан | -269,327 | 6,6 | 0,9 | 1,1 | 1,1 | 1,1 |
| 2,5,5-Триметилгептан | -269,627 | 7,6 | 2,7 | 0,5 | 0,0 | 0,5 |
| 3,3,4-Триметилгептан | -257,727 | -0,9 | 1,8 | 0,6 | 0,8 | 0,6 |
| 3,4,4-Триметилгептан | -257,127 | 1,8 | 1,2 | 0,0 | 1,5 | 0,0 |
| 3,4,5-Триметилгептан | -254,127 | -1,9 | 2,3 | 1,1 | 1,1 | 1,1 |
| 2,2,3,4-Тетраметилгексан | -253,927 | -10,2 | -5,1 | 0,4 | 0,1 | 0,7 |
| 2,2,3,5-Тетраметилгексан | -270,627 | 3,1 | 3,3 | 1,7 | 1,2 | 1,7 |
| 2,2,4,4-Тетраметилгексан | -259,427 | -10,7 | -14,9 | 1,3 | 0,7 | 0,6 |
| 2,2,4,5-Тетраметилгексан | -266,927 | -3,9 | -3,1 | 1,4 | 0,4 | 0,9 |
| 2,3,3,4-Тетраметилгексан | -254,027 | -3,4 | -0,5 | -0,8 | 0,0 | -0,8 |
| 2,3,3,5-Тетраметилгексан | -259,427 | -4,7 | -6,1 | 0,5 | 0,7 | 0,0 |
| 2,3,4,4-Тетраметилгексан | -250,527 | -10,3 | -6,7 | -0,8 | -0,6 | -0,5 |
| 2,3,4,5-Тетраметилгексан | -258,027 | -6,8 | 2,3 | 0,4 | 0,4 | 0,4 |
| 3,3,4,4-Тетраметилгексан | -247,427 | -9,3 | -7,7 | -7,0 | -5,5 | 5,4 |
| Тритретбутилметан | -235,228 | -106,0 | -96,3 | -38,1 | -38,1 | -3,6 |
| 2,2,3,3,4,4,5,5-Октаметилгексан | -248,328 | -104,9 | -55,1 | -52,5 | -48,0 | 0,0 |
| 3,3,4,4-Тетраэтилгексан | -265,528 | -47,0 | -88,3 | -40,5 | -43,9 | -1,5 |
| Среднее абсолютное отклонение для всей выборки |
| 6,2
| 4,5 | 2,7 | 2,5 | 1,5 |
* Соединения, экспериментальные данные для которых использованы при определении парциальных вкладов (табл. 1.10) для уровней 1-3.
Совместной обработкой экспериментальных данных, отмеченных знаком (*) в табл. 1.9, получены величины параметров метода, приведенные в табл. 1.10.
Десять парциальных вкладов (уровень 1), характеризующих все разновидности углерод-углеродных связей в молекулах алканов, дают среднее абсолютное отклонение расчетных величин от экспериментальных значений, равное:
a) 0,10 кДж/моль для линейных алканов;
b) 2,0 кДж/моль - для структур с двумя и меньшим количеством разветвлений при соседних или разделенных двумя связями атомах углерода;
c) 4,5 кДж/моль - для всего объема рассмотренных соединений, в том числе и с тремя или четырьмя последовательно соединенными третичными или четвертичными углеродными атомами, а также с тремя третбутильными заместителями при одном углеродном атоме.
Поскольку средняя абсолютная погрешность расчета ![]() для соединений группы “с” велика, причем для ряда из них она существенно превышает экспериментальную погрешность (0,4-2,0 кДж/моль), естественным шагом при разработке или совершенствовании метода прогнозирования является увеличение глубины его детализации при сохранении общей идеологии.
для соединений группы “с” велика, причем для ряда из них она существенно превышает экспериментальную погрешность (0,4-2,0 кДж/моль), естественным шагом при разработке или совершенствовании метода прогнозирования является увеличение глубины его детализации при сохранении общей идеологии.
В нашем случае это “уровень 2”, на котором аддитивная схема, содержащая десять парциальных вкладов типа (Ci-Cj)1, дополнена десятью парциальными вкладами типа (Ci-Cj)2. Таким образом, для 2-метилбутана и 3,3,4,4-тетраметилгексана имеем:
![]() ,(2-Метилбутана)=2·(C1-C3)1+1·(C1-C2)1+1·(C2-C3)1+2·(C1-C2)2+1·(C1-C1)2+1·(C1-C3)2;
,(2-Метилбутана)=2·(C1-C3)1+1·(C1-C2)1+1·(C2-C3)1+2·(C1-C2)2+1·(C1-C1)2+1·(C1-C3)2;
![]() (3,3,4,4-Тетраметилгексана)=2·(C1-C2)1+2·(C2-C4)1+4·(C1-C4)1+1·(C4-C4)1+2·(C1-C1)2+4·(C1-C2)2+6·(C1-C4)2+2·(C2-C4)2.
(3,3,4,4-Тетраметилгексана)=2·(C1-C2)1+2·(C2-C4)1+4·(C1-C4)1+1·(C4-C4)1+2·(C1-C1)2+4·(C1-C2)2+6·(C1-C4)2+2·(C2-C4)2.
Оптимизации при этом подвергаются все двадцать парциальных вкладов - десять типа (Ci-Cj)1 и десять типа (Ci-Cj)2. Обрабатывается весь массив избранных (*) экспериментальных данных. Результатом расчета явилось следующее. Семь парциальных вкладов - (С1-С1)2, (С1-С2)2, (С1-С3)2, (С1-С4)2, (С2-С2)2, (С2-С3)2, (С3-С3)2 - оказались меньше 0,4 кДж/моль, т.е. являются практически незначимыми, а значит, должны быть исключены из расчета на следующем этапе оптимизации.
Следующий этап оптимизации, направленный на определение взаимно согласованных величин тринадцати парциальных вкладов - десяти первого уровня и трех вкладов второго уровня, показал, что значения всех тринадцати парциальных вкладов (табл. 1.10) превышают наименьшую экспериментальную погрешность определения ![]() . Использование их для прогнозирования энтальпий образования позволяет существенно снизить погрешность расчета. Для всех алканов, приведенных в табл. 1.9, среднее абсолютное отклонение составляет 2,7 кДж/моль. При учете только валентных взаимодействий атомов в молекуле для этой группы веществ имеем 4,5 кДж/моль. Для веществ, использованных при определении парциальных вкладов, погрешность снижается с 2,0 до 0,6 кДж/моль для первого и второго уровней детализации соответственно, что близко к минимальной экспериментальной погрешности.
. Использование их для прогнозирования энтальпий образования позволяет существенно снизить погрешность расчета. Для всех алканов, приведенных в табл. 1.9, среднее абсолютное отклонение составляет 2,7 кДж/моль. При учете только валентных взаимодействий атомов в молекуле для этой группы веществ имеем 4,5 кДж/моль. Для веществ, использованных при определении парциальных вкладов, погрешность снижается с 2,0 до 0,6 кДж/моль для первого и второго уровней детализации соответственно, что близко к минимальной экспериментальной погрешности.
Дальнейшее увеличение глубины детализации аддитивной схемы (“уровень 3”) показал, что восемь из десяти парциальных вкладов типа (Ci-Cj)3 имеют величину меньше 0,4 кДж/моль, т.е. практически незначимы. Дополнение же аддитивной схемы тремя вкладами - (С2-С4)3, (С3-С4)3 и (С4-С4)3 - очень мало меняет точность прогноза. Погрешность расчета составляет 0,10 кДж/моль для “a”, 0,5 кДж/моль для “b” и 2,5 кДж/моль для “c”. Таким образом, при прогнозировании ![]() алканов типа “b” с равным успехом могут использоваться как аддитивная схема Татевского по связям уровня 2, так и уровня 3.
алканов типа “b” с равным успехом могут использоваться как аддитивная схема Татевского по связям уровня 2, так и уровня 3.
При этом обращает на себя внимание тот факт, что ни один из рассмотренных подходов не изменил ситуацию при прогнозировании ![]() алканов с любым количеством разветвлений в молекуле, особенно для структур, изученных в [28].
алканов с любым количеством разветвлений в молекуле, особенно для структур, изученных в [28].
Очевидно, что погрешность в оценках свойства, достигающая нескольких десятков кДж/моль, мало кого может устроить. При этом увеличение глубины детализации рассмотренного метода не дало значимых результатов. Значит, при использовании аддитивных методов для сильно разветвленных структур необходимо рассмотреть иные подходы, позволяющие учесть специфику взаимного расположения групп атомов, ответственных за напряжение в молекуле. Один из таких приемов неоднократно описывался в литературе и состоит в использовании в расчете структурных элементов, представленных несколькими атомами. Принципы классификации структурных элементов, больших, чем единичные атомы, достаточно подробно рассмотрены Татевским в [9].
В соответствии со сказанным схема Татевского по связям, учитывающая только валентные взаимодействия углеродных атомов (уровень 1), была дополнена нами парциальными вкладами для фрагментов молекул, представленных тремя последовательно расположенными атомами углерода. На первом шаге оптимизации рассматривались указанные фрагменты всех разновидностей с применением всего массива экспериментальных данных. В результате было установлено, что значимыми являются только семь парциальных вкладов (табл. 1.10, столбец – “напряженные”) для “троек” атомов с участием четвертичного углеродного атома. Ограниченный объем фактического материала не позволяет в настоящее время обеспечить высокую представительность всех параметров, приведенных в табл. 1.10 для напряженных структур: при определении двух парциальных вкладов участвовали по одному веществу и двух - по два вещества. Тем не менее следует признать, что все параметры взаимно непротиворечивы. Их применение позволяет снизить среднее абсолютное отклонение в расчете с 2,5 кДж/моль до 1,5 кДж/моль для всего объема рассмотренных соединений. Для той же выборки веществ метод Татевского с набором параметров в редакции [11] дает среднюю погрешность в оценках, равную 2,8 кДж/моль, метод Бенсона в редакции [5] – 6,2 кДж/моль.
Таким образом, на рассмотренном примере мы попытались изложить некоторые важные аспекты анализа и совершенствования аддитивных методов, уточнили значения параметров одной из аддитивных схем, показали ее лучшую по сравнению с методом Бенсона работоспособность в приложении к алканам и обозначили проблему прогнозирования ![]() алканов, имеющих значительные напряжения в молекуле. Очевидно, что такие же проблемы возникнут и при расчете энтальпий образования соединений других классов с сильно разветвленными алкильными заместителями. Аналогичный подход использован нами для некоторых технически важных веществ других классов органических соединений. Результаты представлены ниже с необходимым комментарием.
алканов, имеющих значительные напряжения в молекуле. Очевидно, что такие же проблемы возникнут и при расчете энтальпий образования соединений других классов с сильно разветвленными алкильными заместителями. Аналогичный подход использован нами для некоторых технически важных веществ других классов органических соединений. Результаты представлены ниже с необходимым комментарием.
Алкилбензолы и их функциональные производные
Объем экспериментальных сведений для ![]() ароматических углеводородов существенно меньше, чем для алканов, и строение молекул соединений, для которых имеются надежные калориметрические данные, не отличается большим разнообразием. Это не позволяет в настоящее время выработать подходы к прогнозированию их энтальпий образования, опираясь только на калориметрические данные. Для этой цели нами использована вся совокупность фактического материала и возможности неэмпирических методов расчета энергии и геометрии молекул, а также метода молекулярной механики с силовым полем Эллинджера.
ароматических углеводородов существенно меньше, чем для алканов, и строение молекул соединений, для которых имеются надежные калориметрические данные, не отличается большим разнообразием. Это не позволяет в настоящее время выработать подходы к прогнозированию их энтальпий образования, опираясь только на калориметрические данные. Для этой цели нами использована вся совокупность фактического материала и возможности неэмпирических методов расчета энергии и геометрии молекул, а также метода молекулярной механики с силовым полем Эллинджера.
В результате можно с уверенностью говорить о том, что при использовании любого аддитивного метода для ![]() алкилароматических углеводородов необходимо вводить поправки, природа которых в основном имеет стерическое происхождение. Величина этих поправок никоим образом не является постоянной, как это принято, например, в методе Бенсона, а зависит от эффективных размеров взаимодействующих групп, от количества рядом расположенных заместителей и от их взаимной ориентации.
алкилароматических углеводородов необходимо вводить поправки, природа которых в основном имеет стерическое происхождение. Величина этих поправок никоим образом не является постоянной, как это принято, например, в методе Бенсона, а зависит от эффективных размеров взаимодействующих групп, от количества рядом расположенных заместителей и от их взаимной ориентации.
Последовательное применение метода Татевского по связям к накопленным к настоящему времени экспериментальным данным позволило определить значения парциальных вкладов, которые приведены в табл. 1.11. Все парциальные вклады получены на весьма представительных выборках и могут считаться достаточно надежными, чтобы быть рекомендованными к применению.
В отношении поправок (табл. 1.11) необходимо отметить, что большинство из них определено по одному-двум источникам экспериментальной информации. Однако все приведенные значения прошли дополнительное тестирование неэмпирическими методами расчета. Выполненный нами анализ полученных при этом результатов показал, что использование аддитивных подходов на этапе введения поправок для ![]() алкилароматических соединений, имеющих три и большее количество алкильных заместителей в молекуле, может рассматриваться лишь в качестве первого приближения. Недостаточно конструктивен, на наш взгляд, также подход, предложенный в свое время Коксом и Пилчером [2] для полизамещенных бензолов и состоящий в дополнении орто-эффектов поправками, учитывающими тройное взаимодействие заместителей в молекуле.
алкилароматических соединений, имеющих три и большее количество алкильных заместителей в молекуле, может рассматриваться лишь в качестве первого приближения. Недостаточно конструктивен, на наш взгляд, также подход, предложенный в свое время Коксом и Пилчером [2] для полизамещенных бензолов и состоящий в дополнении орто-эффектов поправками, учитывающими тройное взаимодействие заместителей в молекуле.
Похожие работы
... изменение теплоемкости. Дж/моль*К Из правой части выражаем: Задание №5 Для первого соединения рассчитать плотность вещества при температуре 730 К и давлении 100 бар. Определить фазовое состояние компонента. Для определения плотности вещества воспользуемся методом прогнозирования плотности индивидуальных веществ с использованием коэффициента сжимаемости. где -плотность вещества; М- ...

... идеально-газовым энтальпиям образования при других температурах или к , т.е. к свойству вещества в реальном состоянии. Следует признать, что из всего многообразия аддитивных схем для прогнозирования энтальпий образования органических веществ метод Бенсона в течение продолжительного периода применяется наиболее широко. Объясняется это, вероятно, тем, что этим методом охвачен наиболее широкий круг ...
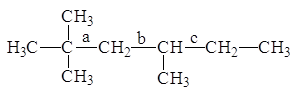
... по связям. Прогнозирование органических соединений методом Бенсона по атомам с их первым окружением Следует признать, что из всего многообразия аддитивных схем для прогнозирования энтальпий образования органических веществ метод Бенсона в течение продолжительного периода применяется наиболее широко. Объясняется это, вероятно, тем, что этим методом охвачен наиболее широкий круг соединений. Для ...
... Задание №5 Для первого соединения рассчитать плотность вещества при температуре 730 К и давлении 100 бар. Определить фазовое состояние компонента. Для определения плотности вещества воспользуемся методом прогнозирования плотности индивидуальных веществ с использованием коэффициента сжимаемости. где -плотность вещества; М- молярная масса; V-объем. Для данного вещества найдем коэффициент ...
0 комментариев