Навигация
Сульфосалициловая кислота, 10%-ный раствор, или сульфосалицилат натрия, насыщенный раствор
86506
знаков
28
таблиц
16
изображений
1. Сульфосалициловая кислота, 10%-ный раствор, или сульфосалицилат натрия, насыщенный раствор.
2. Аммиак, разбавленный раствор. Смешивают 200 мл концентрированного аммиака и 300 мл дистиллированной воды.
3. Стандартный раствор соли железа. Растворяют 0,8634 г железоаммонийных квасцов в дистиллированной воде, к раствору добавляют 10 мл H2SO4 плотностью 1,84 г/см3, разбавляют в мерной колбе на 1 л. Отбирают 100 мл полученного раствора, разбавляют водой в мерной колбе снова до 1 л.
В 1 мл раствора содержится 0,01 мг железа.
Ход определения общего содержания железа.
В коническую колбу вместимостью 50 мл наливают 10 мл анализируемой воды. В этом объеме должно содержаться от 1 до 10 мкг железа, что соответствует концентрациям от 0,1 до 1 мг/л. Более концентрированные по содержанию железа сточные воды предварительно разбавляют в мерной колбе так, чтобы содержание железа в 10 мл полученного раствора было в указанных пределах. Затем в пробирку приливают 5 мл раствора сульфосалициловой кислоты и 5 мл раствора аммиака.
Измеряют оптическую плотность полученного раствора на длине волны 420-430 нм по отношению к холостому раствору. Молярный коэффициент поглощения равен 5,5103.
Содержание железа находят по градуировочному графику, для построения которого наливают из микробюретки 1-10 мл стандартного раствора, разбавляют до 10 мл дистиллированной водой и продолжают как при анализе пробы.
Построение градуировочного графика.
Были приготовлены реактивы как указано в методике. Взяли 2,0; 4,0; 6,0; 8,0; 10,0 мл стандартного раствора. Фотометрировали на ФЭКе на длине волны 440 нм в кюветах 3 см. Данные приведены в таблице 2.5.1.
Таблица 2.5.1.
Зависимость оптической плотности растворов от концентрации железа| С, мкг/мл | 0,4 | 0,8 | 1,2 | 1,6 | 2,0 |
| D1 | 0,075 | 0,17 | 0,26 | 0,36 | 0,44 |
| D2 | 0,075 | 0,16 | 0,25 | 0,36 | 0,44 |
| D3 | 0,080 | 0,17 | 0,26 | 0,35 | 0,44 |
Статистическая обработка проводилась на ЭВМ по методу наименьших квадратов. Получено уравнение регрессии:
y=2,310-1x
rт=0,878, rр=1,000
b=2,310-11,310-2.
Рисунок 2.5.1.
Г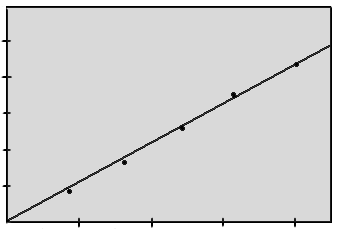
D
0,5
0,4
0,3
0,2
0,1
радуировочный график поглощения растворов железа0,5 1,0 1,5 2,0 С, мкг/мл
2.5.2. Фотометрическое определение алюминия с алюминоном [40]
Метод основан на способности иона Al3+ образовывать с алюминием красное труднорастворимое комплексное соединение.
Реакция осуществляется при pH=4,9 в присутствии сульфата аммония. Оптическую плотность измеряют на длине волны 540 нм. Чувствительность метода 0,05 мг/л алюминия.
1.Приготовление основного стандартного раствора с концентрацией 0,10 г/мл. Стандартный раствор можно приготовить из алюминиевой стружки. Для этого 0,1 г стружки растворяют в 5 мл концентрированной соляной кислоты при нагревании и перемешивании. Переливают раствор в мерную колбу на 1000 мл и доводят до метки дистиллированной водой.
2.Приготовление рабочего стандартного раствора с концентрацией 0,005 мг/мл. 5,0 мл основного стандартного раствора помещают в мерную колбу на 100 мл и доводят объем до метки дистиллированной водой. Раствор готовят в день проведения анализа.
3.Приготовление раствора сульфата аммония. 50,0 г сульфата аммония растворяют в 100 мл дистиллированной воды.
4.Приготовление ацетатного буфера (pH=4,91). 241 г CH3COONa помещают в колбу на 1000 мл и растворяют в 500 мл дистиллированной воды. Дают раствору остыть, добавляют 155 мл ледяной уксусной кислоты, доводят объем до метки дистиллированной водой. pH буфера устанавливают потенциометрически и, при необходимости, корректируют, добавляя NaOH или CH3COOH. Для анализа исходный буферный раствор разбавляют в 10 раз дистиллированной водой. Срок хранения 3 месяца.
5. Приготовление раствора алюминона. 0,5 г алюминона растворяют в 125 мл горячей дистиллированной воды, затем добавляют 125 мл разбавленного ацетатного буфера. Раствор готов к употреблению сразу. Хранят его в склянке из темного стекла. Срок хранения 3 месяца.
6. Приготовление раствора гидроокиси натрия. 5,0 г NaOH растворяют в 100 мл дистиллированной воды.
7. Приготовление реакционной смеси. Смешивают одну объемную часть сульфата аммония с двумя частями алюминона и двадцатью двумя – разбавленного ацетатного буфера. В день анализа в необходимом объеме реакционной смеси растворяют аскорбиновую кислоту по 25 мг на каждые 25 мл реакционной смеси. Раствор можно хранить в течение 1 месяца.
Проведение анализа.
Проведению анализа может помешать окисное железо (Fe3+). Влияние железа массовой концентрации 0,3 мг/л и более устраняется восстановлением его аскорбиновой кислотой до закисного железа (Fe2+).
В мерную колбу на 50 мл, где уже находится исследуемый раствор, добавляют 25 мл реакционной смеси, 25-30 мг аскорбиновой кислоты, доводят до метки дистиллированной водой. Раствор выдерживают в течении 25-30 мин. Затем измеряют его оптическую плотность на длине волны 540 нм в кювете с толщиной слоя 30 мм. Измерения проводят относительно холостой пробы. Для приготовления холостой пробы в мерную колбу на 50 мл помещают 25 мл реакционной смеси, 25-30 мг аскорбиновой кислоты, доводят до метки дистиллированной водой. Количество алюминия в пробе находят по градуировочному графику. При необходимости производят разбавление исследуемого раствора.
Построение градуировочного графика.
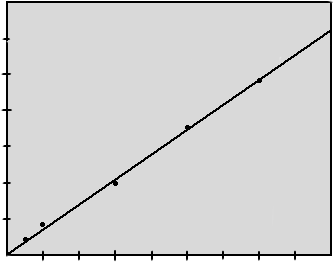
Приготовление реактивов велось по методике. Взяли 0,5; 1,0; 3,0; 5,0; 7,0; 9,0 мл рабочего стандартного раствора. Фотометрировали на длине волны 540 нм в кюветах 3 см. Полученные данные приведены в таблице 2.5.2.
Таблица 2.5.2.
Зависимость оптической плотности растворов от концентрации алюминия
| С, мкг/мл | 0,05 | 0,1 | 0,3 | 0,5 | 0,7 | 0,9 |
| D1 | 0,04 | 0,08 | 0,22 | 0,36 | 0,50 | 0,66 |
| D2 | 0,03 | 0,07 | 0,20 | 0,34 | 0,48 | 0,64 |
| D3 | 0,04 | 0,07 | 0,21 | 0,34 | 0,48 | 0,64 |
Статистическая обработка проводилась на ЭВМ с помощью метода наименьших квадратов. Получено уравнение регрессии:
y=6,910-1x
rт=0,878, rр=1,000
b=6,910-11,210-2.
Рисунок 2.5.2.
Градуировочный график поглощения растворов алюминия
D
0,6
0,5
0,4
0,3
0,2
0,1
0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 С, мкг/мл
2.5.3. Фотометрическое определение ванадия [41]
1. Приготовление эталонного раствора ванадия 1000 мг/л.
В стакане вместимостью 100 мл растворяют 0,5740 г метаванадиевокислого аммония в 40 мл разбавленной азотной кислоты (1:4), количественно переносят в мерную колбу вместимостью 250 мл и доводят до метки дистиллированной водой.
2. Приготовление рабочего эталонного раствора ванадия 50 мг/л.
5 мл эталонного раствора ванадия 1000 мг/л количественно переносят в мерную колбу вместимостью 100 мл и доводят до метки дистиллированной водой.
3. Приготовление раствора вольфрамовокислого натрия 185 г/мл.
В стакане вместимостью 50 мл растворяют 18,5 г двуводного вольфрамовокислого натрия (Na2WO42H2O) в горячей дистиллированной воде, охлаждают до комнатной температуры, количественно переносят в мерную колбу вместимостью 100 мл и доводят до метки дистиллированной водой.
Проведение анализа.
В стакан вместимостью 100 мл помещают аликвоту раствора в зависимости от ожидаемого содержания ванадия, 2 мл разбавленной серной кислоты (1:1), 5 мл разбавленной ортофосфорной кислоты и 2,5 мл вольфрамовокислого натрия. Каждый раз содержимое стакана перемешивают встряхиванием. Раствор нагревают до 40-70C, охлаждают до комнатной температуры, количественно переносят в мерную колбу вместимостью 50 мл, доводят до метки дистиллированной водой. Раствор должен быть прозрачным. Выдерживают в течение 60 мин. Затем измеряют его оптическую плотность на длине волны 440 нм в кювете с толщиной слоя 5 см. Измерения проводят относительно холостой пробы. Для приготовления холостой пробы в мерную колбу на 50 мл помещают 2 мл разбавленной серной кислоты (1:1), 5 мл разбавленной ортофосфорной кислоты и 2,5 мл вольфрамовокислого натрия, доводят до метки дистиллированной водой. Количество ванадия находят по градуировочному графику.
Построение градуировочного графика.
Приготовление растворов велось по методике. Взяли 1,0; 2,0; 4,0; 6,0; 8,0; 10,0 мл рабочего эталонного раствора. Фотометрировали на длине волны 440 нм в кюветах 5 см. Полученные данные приведены в таблице 2.5.3.
Таблица 2.5.3.
Зависимость оптической плотности растворов от концентрации ванадия
| С, мкг/мл | 1,0 | 2,0 | 4,0 | 6,0 | 8,0 | 10,0 |
| D1 | 0,06 | 0,12 | 0,26 | 0,39 | 0,50 | 0,62 |
| D2 | 0,05 | 0,12 | 0,26 | 0,38 | 0,50 | 0,62 |
| D3 | 0,05 | 0,11 | 0,25 | 0,38 | 0,50 | 0,60 |
Статистическая обработка проводилась на ЭВМ с помощью метода наименьших квадратов. Получено уравнение:
y=6,310-2x
rт=0,811, rр=0,999
b=6,310-23,110-3.
Рисунок 2.5.3.
Г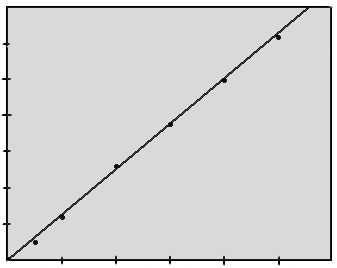
D
0,6
0,5
0,4
0,3
0,2
0,1
радуировочный график поглощения растворов ванадия2,0 4,0 6,0 8,0 10,0 С, мкг/мл
2.5.4. Фотометрическое определение галлия [42]Метод основан на образовании ионного ассоциата хлоргаллата родамина B и измерении оптической плотности его экстрактов. Интенсивная красно-фиолетовая окраска экстракта обеспечивает чувствительный способ определения малых количеств галлия. В этих условиях аналогично реагируют Sb (V), Au (III), Tl (III) и Fe (III). Вольфрам (VI) образует вольфрамат родамина B, не растворимый в толуоле.
Al, Zn, Cu, Ta и In не дают окраски при экстракции толуолом. Окислители, в том числе нитраты, должны отсутствовать в растворе. Сурьма и другие перечисленные металлы не реагируют с родамином B, если они восстановлены до более низких степеней валентности [Sb(III), Au(0), Tl(I), Fe(II)].
Реактивы:
1. Кислота соляная, 6-н. раствор.
2. Титан треххлористый, раствор, разбавленный концентрированной соляной кислотой 1:1.
3. Родамин B, 0,5%-ный раствор в 6-н. растворе соляной кислоты.
4. Смесь толуола с бутилацетатом. Готовят смешением толуола с бутилацетатом в соотношении 2:1.
5. Галлий металлический.
Приготовление стандартных растворов галлия.
1. Раствор А. Навеску галлия массой 0,1 г растворяют в 10 мл 6-н. раствора соляной кислоты при нагревании с добавлением нескольких капель пергидроля. Полученный раствор выпаривают досуха. Остаток снова растворяют в 6-н. растворе соляной кислоты, раствор переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора А содержит 1000 мкг галлия.
2. Раствор Б. Отбирают пипеткой 10 мл раствора А и переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора Б содержит 100 мкг галлия.
3. Раствор В. Отбирают пипеткой 10 мл раствора Б и переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора В содержит 10 мкг галлия.
4. Раствор Г. Отбирают пипеткой 10 мл раствора В и переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора Г содержит 1 мкг галлия.
Проведение анализа.
В мерную колбу вместимостью 50 мл (или делительную воронку вместимостью 25 мл) отбирают 5 мл анализируемого раствора и прибавляют по каплям при помешивании 1 мл треххлористого титана. Через 5 мин в колбу (или воронку) прибавляют 0,4 мл 0,5%-ного раствора родамина B, перемешивают и выдерживают в течение 5 мин. Затем приливают 5 мл смеси толуола с бутилацетатом, закрывают колбу (или воронку) притертой пробкой и встряхивают 2 мин. После разделения слоев осторожно сливают в кювету верхний органический слой, окрашенный в красно-фиолетовый цвет, и закрывают кювету крышкой. Колориметрирование производят на длине волны 540 нм в кювете с толщиной слоя 5 мм.
Нулевой раствор готовят следующим способом: в мерную колбу вместимостью 50 мл (или делительную воронку вместимостью 50 мл) наливают 10 мл 6-н. раствора соляной кислоты, приливают 2 мл треххлористого титана, 0,8 мл 0,5%-ного раствора родамина B, 10 мл смеси толуола с бутилацетатом, закрывают притертой пробкой колбу (или воронку) и встряхивают в течение 2 мин. После разделения органический слой сливают в кюветы для колориметрирования.
Построение градуировочного графика.
Приготовление реактивов велось по методике. Взяли 1,0; 2,0; 3,0; 4,0; 5,0 мл стандартного раствора Г. Фотометрировали на длине волны 540 нм в кюветах 5 мм. Данные приведены в таблице 2.5.4.
Таблица 2.5.4.
Зависимость оптической плотности растворов от концентрации галлия
| M, мкг | 2 | 4 | 6 | 8 | 10 |
| D1 | 0,09 | 0,13 | 0,19 | 0,26 | 0,27 |
| D2 | 0,10 | 0,14 | 0,18 | 0,26 | 0,30 |
| D3 | 0,11 | 0,14 | 0,18 | 0,23 | 0,29 |
Статистическая обработка проводилась на ЭВМ с помощью метода наименьших квадратов. Получено уравнение регрессии:
y=3,710-2+2,510-2x
rт=0,878, rр=0,983
a=3,710-25,710-2
b=2,510-28,610-3
Рисунок 2.5.4.
Г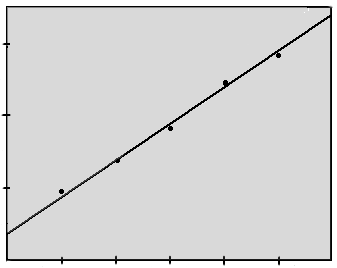
D
0,3
0,2
0,1
радуировочный график поглощения растворов галлия2 4 6 8 10 m, мкг
2.5.5. Методика определения кремния [43].
При растворении плава в соляной кислоте выделяется кремниевая кислота:
Na2SiO3+2HCl2NaCl+H2SiO3
Некоторая часть образовавшейся кремниевой кислоты остается в растворе в виде гидрозоля. Чтобы перевести ее полностью в осадок, раствор плава выпаривают и оcтаток от выпаривания высушивают, при этом золь кремниевой кислоты переходит в гель. При прокаливании до 1000C кремнекислота теряет воду, превращаясь в кремниевый ангидрид:
H2SiO3![]() SiO2+H2O
SiO2+H2O
Выполнение определения.
Раствор в фарфоровой чашке после выщелачивания плава разбавляют 10%-ным раствором соляной кислоты до объема 150 мл и выпаривают досуха на водяной бане (избегать разбрызгивания). Затем добавляют 10 мл соляной кислоты (пл. 1,19) и снова выпаривают на водяной бане досуха. После этого сухой остаток сушат 1 ч в сушильном шкафу при температуре 120C. Более эффективна сушка на водяной бане до полного удаления запаха HCl с последующим прокаливанием в течение 2 ч.
Содержимое чашки охлаждают и осторожно смачивают 15 мл HCl (пл. 1,19), прибавляя ее по каплям. Покрывают чашку часовым стеклом и оставляют стоять 10 мин. После этого содержимое чашки обрабатывают 70-80 мл горячей дистиллированной воды, дают стоять 10 мин на кипящей водяной бане и отстоявшийся раствор фильтруют через фильтр с красной лентой, не перенося осадка на фильтр. Осадок в чашке промывают горячей водой, вначале декантацией, затем переносят его на фильтр и промывают его на фильтре до исчезновения реакции на хлор с азотнокислым серебром в промывных водах.
Для удаления невидимых на белом фоне чашки оставшихся крупинок кремнекислоты чашку протирают маленьким кусочком фильтровальной бумаги, который присоединяют к осадку (№1) на фильтре. Фильтрат количественно переносят в ту же чашку, в которой проводилось выпаривание, и вторично выпаривают досуха и до удаления запаха HCl. После охлаждения сухой остаток в чашке смачивают по каплям соляной кислотой (пл. 1,19) в количестве 5-7 мл, накрывают часовым стеклом и оставляют стоять 10 мин. Затем содержимое чашки обрабатывают 30 мл горячей дистиллированной воды и дают стоять 10 мин на кипящей водяной бане и далее поступают так же, как и в первый раз: отфильтровывают, промывают и т. д.
Вторичной обработкой фильтрата достигают более полного перевода золя кремнекислоты в осадок. Фильтрат №1 сохраняют для дальнейшего анализа, а осадок №2 на фильтре присоединяют вместе с фильтром к ранее выделенному осадку №1 кремнекислоты.
Общий осадок кремнекислоты вместе с фильтрами озоляют вначале при низкой температуре во взвешенном и прокаленном платиновом или фарфоровом тигле на пламени газовой горелки, помещая тигель на асбестовую сетку, затем на голом огне, постепенно увеличивая пламя. Под конец прокаливают до постоянной массы в муфельной печи (в течение 30-50 мин при t=1000-1100C). Весовая форма SiO2 белого цвета.
2.5.6. Выполнение сплавления и растворения навески силиката (золы) [43]Для сплавления навеску испытуемого материала (золы) в количестве 0,5-1,0 г отвешивают в фарфоровом тигле с точностью до 0,0002 г. К навеске добавляют 4-6 г безводной соды, хорошо растертой в фарфоровой ступке. Величина фарфорового тигля должна быть такой, чтобы он не более чем наполовину был заполнен порошком. Содержимое тигля осторожно и тщательно перемешивают маленьким шпателем, затем смесь сверху засыпают 1-2 г чистой соды.
После этого тигель вносят в разогретую до 200-300C муфельную печь и медленно нагревают до 800-900C в течение 30-40 мин. Затем при данной температуре выдерживают 1,5 ч. После охлаждения тигля плав переносят в фарфоровую чашку емкостью 200-250 мл и растворяют в 30-40 мл HCl (1:1). Тигель несколько раз ополаскивают раствором HCl (1:1), сливая раствор в фарфоровую чашку. Раствор в фарфоровой чашке после выщелачивания сплава выпаривают досуха, избегая разбрызгивания. Затем добавляют 10 мл соляной кислоты (пл. 1,19) и снова выпаривают досуха.
Содержимое чашки охлаждают и осторожно смачивают небольшим количеством HCl (1:1). Оставляют стоять 10 мин, нагревают и отстоявшийся раствор фильтруют, не перенося осадка на фильтр. Осадок в чашке промывают раствором HCl (1:1), вначале декантацией, затем переносят его на фильтр и промывают на фильтре. Полученный раствор анализируют.
2.6. Методики эксперимента 2.6.1. Методика обескремнивания
В стеклянный термостойкий стакан вместимостью 1000 мл помещается 800 мл щелочного раствора концентрацией 200 г/л. Раствор термостатируется до 80C. Затем в стакан помещается 160 г золы и включается механическая мешалка. Время обескремнивания 3 часа.
2.6.2. Методика щелочной и кислотной обработкиОбработка золы проводится в термостойком стакане вместимостью 250 мл. Предварительно раствор едкого натра или серной кислоты термостатируется до необходимой температуры. Затем помещается зола и включается механическая мешалка.
2.6.3. Методика электрохимического выщелачивания
Д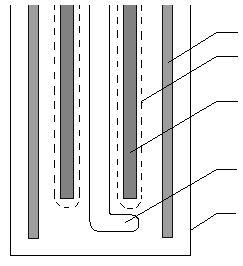
1. Катод
2. Перхлорвиниловая диафрагма
3. Анод
4. Механическая мешалка
5. Химический стакан
ля исследования была собрана электрохимическая ячейка:В электрохимическую ячейку помещается серная кислота или раствор едкого натра объемом 200-250 мл и концентрацией 100-400 г/л и, при необходимости, термостатируется. После этого в ячейку помещается навеска (20-25 г) обескремненной золы, включается механическая мешалка и электролизер. Сила тока задается в пределах 1-10 А.
3. ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ 3.1. Отработка методики анализа на содержание ванадияВанадий определялся по методике ГОСТ 10364-90. Она предусмотрена для определения ванадия (V). Но в наших опытах при электролизе ванадийсодержащих растворов ванадий может перейти в более низкую степень окисления (III, IV). Поэтому нами предварительно отрабатывалась методика окисления ванадия.
Опыты проводились с модельными растворами NH4VO3, которые готовились следующим образом. Навеска NH4VO3 массой 0,5740 г растворялась в растворе H2SO4 (1:1), и определялось содержание ванадия по методике. Оно составило 50,95,8 мг/л (статистическая обработка в приложении 1).
Затем этот раствор восстанавливали цинком при кипячении в течение 0,5 ч до V(II) фиолетовой окраски. Анализ показал содержание ванадия 32,51,6 мг/л, что дает заниженные результаты.
Для окисления V(II) до V(V) использовалась концентрированная азотная кислота в расчете 5 мл HNO3 на 100 мл раствора. Анализ окисленного раствора по методике показал значение 49,92,3 мг/л.
Таким образом, предварительное окисление аликвоты азотной кислотой позволяет определить истинное содержание ванадия в растворе.
3.2. Поведение галлия и ванадия при выщелачивании
Предварительными исследованиями было показано, что обработка золы ТЭЦ раствором щелочи 200 г/л при температуре 80C и времени выщелачивания 2 ч приводит к извлечению 49,7% кремния, 5,5% алюминия и 5,6% ванадия. То есть такая обработка приводит к концентрированию металлов в зольном остатке. Удаление аморфной части SiO2, частичное разрушение частиц золы должно обеспечить более эффективное извлечение галлия и ванадия из золы. Таким образом обработанная зола использовалась в последующих опытах.
3.2.1. Исследование влияния температуры и добавки NaCl на эффективность выщелачивания галлия, ванадия, железа и алюминия в сернокислой средеИсследование проводилось при условиях:
концентрация H2SO4
0 комментариев