Навигация
Реакции с разрывом С¾O связи
48883
знака
13
таблиц
0
изображений
2. Реакции с разрывом С¾O связи.
Образование галогенидов.
При действии неорганических галогенангидридов на третичные и вторичные спирты происходит в основном обмен гидроксила на галоген:
3(CH3)3COH + PBr3 → 3(CH3)3CBr + P(OH)3
Обмен гидроксила на галоген происходит и при действии PBr3 и PI3 на первичные спирты:
3C2H5OH + PBr3 → 3C2H5Br + P(OH)3
При действии галогенводородных кислот на спирты также образуются алкилгалогениды.
Реакция может протекать либо по механизму SN2, либо по SN1. Например:
![]()
Br-
RCH2OH + H+ → R¾CH2 ¾O+¾O → RCH2Br + H2O SN2
½
H для первичных спиртов
![]()
![]()
![]()
![]() R R -H2O R Br- R
R R -H2O R Br- R
![]()
![]() R’¾C¾OH + H+ → R’¾C¾O+¾H R’¾C+ → R’¾C¾Br SN1
R’¾C¾OH + H+ → R’¾C¾O+¾H R’¾C+ → R’¾C¾Br SN1
![]()
![]()
![]()
![]() R” R” ½
R” R”
R” R” ½
R” R”
H для вторичных и третичных спитртов
Для успешной замены гидроксильной группы на хлор используют реактив Лукаса (соляная кислота + ZnCl2 ). Реакционная способность спиртов в этих реакциях изменяется в ряду: третичные>вторичные>первичные.
3. Реакции с участием группы OH и атома водорода, стоящего у соседнего атома углерода.
Дегидратация спиртов в олефины. Все спирты (кроме метилового) при пропускании их паров над нагретой до ~375°С окисью алюминия отщепляют воду и образуют олефин:
Al2O3
![]() СН3─СН2ОН
СН2=СН2 + Н2О
СН3─СН2ОН
СН2=СН2 + Н2О
Особенно легко элиминируется вода из третичных спиртов.
Дегидрогенизация. Образование разных продуктов в реакциях дегидрогенизации и окисления является важнейшим свойством, позволяющим отличить первичные, вторичные и третичные спирты.
При пропускании паров первичного или вторичного, но не третичного спирта над металлической медью при повышенной температуре происходит выделение двух атомов водорода, и спирт превращается в альдегид:
Cu
RCH2OH → R−C−H + H2
200-300 °C ║
O
Вторичные спирты дают в этих условиях кетоны:
R
\ Cu
CHOH → R’−C−R + H2
/ 200-300 °C ║
R’ O
Окисление. Для окисления спиртов обычно используют сильные окислители: KMnO4, K2Cr2O7 и H2SO4. При окислении первичных спиртов образуются альдегиды, которые далее могут окисляться до карбоновых кислот:
RCH2OH + [O] → R─C─H + H2O
║
O
R
\
CHOH + [O] → R’−C−R + H2O
/ ║
R’ O
Вторичные спирты при окислении превращаются в кетоны:
OH O
½ [O] ║
CH3CHCH3 → CH3CCH3
Пропанол-2 пропанон-2
Третичные спирты значительно труднее окисляются, чем первичные и вторичные, причём с разрывом связей C¾C(OH):
(а) O O CH3
║ ║ ½
![]()
![]()
![]() H¾C¾OH + CH3CH2C¾CHCH3
H¾C¾OH + CH3CH2C¾CHCH3
Муравьиная к-та 2-метилпентанон-3
CH3 O O CH3
½ [O] (б) ║ ║ ½
![]() CH3CH2 ¾ C¾OH CH3 ¾C¾OH + CH3C¾CHCH3
CH3CH2 ¾ C¾OH CH3 ¾C¾OH + CH3C¾CHCH3
½ Уксусная к-та 2-метилбутанон-3
CH3CHCH3
2,3-диметилпентанон-3 O O
(в) ║ ║
CH3CCH3 + CH3CH2CCH3
Ацетон бутанон-2
Двухатомные спирты, или гликоли (алкандиолы)
Двугидроксильные производные алканов (открыты Вюрцем) носят название гликолей или алкандиолов. Гидроксилы в алкандиолах находятся либо при соседних, либо более удалённых друг от друга углеродных атомах. 1,2-Гликоли имеют сладкий вкус, откуда и происходит название класса. Низшие гликоли – смешивающиеся с водой вязкие жидкости большей плотности, чем одноатомные спирты. Кипят при высокой температуре. Гликоли с короткой углеродной цепью, и прежде всего этиленгликоль, не растворяются в углеводородах и эфире, но смешиваются с водой и спиртами; как растворители они ближе стоят к воде и метанолу, чем к обычным органическим растворителям.
Способы получения
В принципе гликоли могут быть получены всеми синтетическими способами получения спиртов.
Гидролиз дигалогенпроизводных:
ClCH2─CH2Cl + 2H2O → HOCH2─CH2OH + 2HCl
или
ClCH2─CH2OH + H2O → HOCH2─CH2OH + HCl
Восстановление сложных эфиров двухосновных кислот:
O O
║ ║
C2H5O─C─(CH2)n─C─OC2H5 + 8Na+6C2H5OH → HOCH2─(CH2)n─CH2OH +8C2H5ONa
3CH2=CH2 + 4H2O + 2KMnO4 → 3HOCH2─CH2OH + 2KOH + 2MnO2
Получение гликолей через хлоргидрины. Действием хлора и воды на олефин можно получить хлоргидрин, например ClCH2─CH2OH. Хлоргидрин может быть превращён гидролизом непосредственно в гликоль.
Пинаконы получают восстановлением (неполным) кетонов электрохимически или действием магния в присутствии иода:
СH3 H3C CH3 H3C CH3
½ | | 2H2O | |
2 C=O + 2Mg + I2 → CH3─C─C─CH3 → CH3─C─C─CH3
½ | | | |
CH3 IMgO OMgI HO OH
Бутандиол-1,4 (важный продукт, являющийся промежуточным продуктом при получении бутадиена и далее синтетического каучука) получают в промышленности гидрированием бутин-2-диола-1,4 (НОН2С─С≡С─СН2ОН).
В промышленности этиленгликоль синтезируют из окида этилена, который получают окислением этилена:
250 °C H2O
![]()
![]() 2CH2=CH2 +O2 Ag CH2¾CH2H+ HOCH2CH2OH
2CH2=CH2 +O2 Ag CH2¾CH2H+ HOCH2CH2OH
\ /
O
a-окись
Химические свойства гликолей
Так же как и одноатомные спирты, гликоли могут иметь первичные, вторичные и третичные гидроксилы. Этиленгликоль – двупервичный спирт, пропиленгликоль – первично-вторичный, пинакон – двутретичный. Всё сказанное о свойствах первичных, вторичных и третичных спиртов приложимо и к соответствующим гликолям.
1. Гликоли легко образуют хлорангидриды и бромгидрины при действии HCl или HBr, но второй гидроксил замещается на галоген труднее (лучше действием PCl5 или SOCl2).
2. При действии кислот гликоли дают два ряда сложных эфиров:
O O O
║ ║ ║
HOCH2─CH2─O─C─R R─C─O─CH2─CH2─O─C─R
3. При окислении первичных гликолей образуются альдегиды. Так, окислением этиленгликоля получают глиоксаль:
[O] [O]
HOCH2─CH2OH → HOCH2─C=O → O=C─C=O
½ │ │
H H H
4. Дегидратация гликолей (кислотами или хлористым цинком) приводит к образованию альдегидов (или кетонов). Считают, что механизм этой дегидратации состоит в том, что сначала путём отрыва одной гидроксильной группы протоном образуется карбониевый катион, а затем атом водорода вместе со своей парой электронов (в виде гидрид-иона) перемещается к карбониевому углероду (гидридное перемещение):
H
+ │
CH2─CH2 → CH2─CH → CH3─CH + H+
│ │ │ ║
H+ OH OH O O
│
H
При дегидратации пинаконов мигрирует не водород, а метильная группа и происходит пинаколиновая перегруппировка, сопровождающаяся изменением углеродного скелета:
СН3 СН3 СН3 СН3 СН3
│ │ ½ │ │
СН3─C ¾С─СН3 → С+─С─СН3 → СН3─С ─ С─СН3 + Н+
│ │ ½ ½ │ ║
ОН ОН СН3 О СН3О
Н+ │ пинаколин
пинакон Н
5. Альдегиды в кислой среде ацетилируют 1,2-гликоли, образуя циклические ацетали (в кислой, но не щелочной среде в результате гидролиза ацеталя регенерируются исходные вещества):
![]() СН2─О
СН2─О
![]() СН2─ОН Н+
СН2─ОН Н+
![]()
![]() │ + О=С─СН3 С─СН3
│ + О=С─СН3 С─СН3
![]() СН2─ОН
│ Н+, Н2О
СН2─ОН
│ Н+, Н2О
Н СН2─О
ацеталь
1,3-Гликоли способны реагировать подобным образом, давая шестичленные циклические ацетали.
Для осуществления реакций ацетилирования необходима возможность приведения обоих гидроксилов в одну плоскость, т.е. возможность свободного вращения вокруг углерод-углеродной связи:
НО─С
│
С─ОН
Это условие соблюдается у гликолей с открытой цепью, но не всегда у циклических.
Многоатомные спирты
Трёхатомные спирты – алкантриолы
Единственным важным представителем алкантриолов является глицерин (пропантриол-1,2,3). Это очень вязкая бесцветная сладкая жидкость; т. пл. 17°С, т. кип. 290°С.
Глицерин был получен гидролизом жиров, которые являются сложными эфирами глицерина и высших гомологов уксусной кислоты (и их олефиновых изологов). При гидролизе жиров перегретым паром глицерин остаётся в водном растворе, который отделяют от слоя расплавленных жирных кислот; после отгонки воды из этого раствора может быть выделен глицерин.
Некоторое количество глицерина образуется при брожении сахаров.
В настоящее время осуществлён промышленный синтез глицерина из пропилена, выделяемого из газов крекинга нефти. Этот синтез является доказательством строения глицерина как пропантриола.
Сначала путём хлорирования пропилена при высокой температуре (500°С) получают хлористый аллил, сохраняющий двойную связь (реакция Львова):
СН2=СН─СН3 + Сl2 → CH2=CH─CH2Cl + HCl
Затем присоединением хлора и воды хлористый аллил превращают в 1,3-дихлорпропанол-2
Cl OH Cl
│ │ │
CH2=CH─CH2Cl + Cl2 + H2O → CH2─CH─CH2 + HCl
гидролиз которого даёт глицерин:
Cl OH Cl ОН ОН ОН
│ │ │ │ │ │
CH2─CH─CH2 + 2Н2О → CH2─CH─CH2 + 2HСl
1,3-дихлорпропанол-2 пропантриол-1,2,3
(глицерин)
Глицерин даёт с кислотами три ряда сложных эфиров: моно-, ди- и триэфиры. Для первых и вторых возможны изомеры: продукты этерификации по первичным и вторичным группам. При действии HCl на глицерин получается смесь двух монохлоргидринов глицерина, содержащая больше α-монохлоргидрина СН2ОН─СНОН─СН2Cl и меньше β-изомера СH2OH─CHCl─CH2OH. При обработке щёлочью оба изомера дают один и тот же глицидный спирт
Н2С─СН─СН2ОН
\ /
О
При обработке глицерина хлористым водородом в более жёстких условиях образуются два дихлоргидрина
СН2Cl─СНОН─СН2Cl СH2OH─CHCl─CH2Cl
при обработке щёлочью дающие эпихлоргидрин глицерина
Н2С─СН─СН2Сl
\ /
О
Являясь одновременно первичным и вторичным спиртом, глицерин, нашедший многообразное применение в органическом синтезе, при окислении образует смесь соответствующего альдегида и кетона:
![]()
![]()
![]() СН2ОН─СНОН─С=О
СН2ОН─СНОН─С=О
|
Н
![]() СH2OH─CНОН─CH2OH
Глицериновый альдегид
СH2OH─CНОН─CH2OH
Глицериновый альдегид
СH2OH─CО─CH2OH
диоксиацетон
Диоксиацетон может быть получен хлорированием ацетона в 1,3-дихлорацетон СH2Cl─CО─CH2Cl и гидролизом последнего. Эта реакция также подтверждает строение глицерина.
Четырёхатомные, пятиатомные и шестиатомные спирты (эритриты, пентиты и гекситы)
Эритрит (бутантетраол-1,2,3,4) встречается в свободном виде и в виде сложных эфиров в водорослях и некоторых плесенях. Синтетический четырёхатомный спирт эритрит был получен из бутадиена СH2=СH─CН=CH2 следующим путём:
O O
║ ║
CН=CH2 +Br2 CH─CH2Br 2AgO CCH3 CH─CH2─OCCH3 +Br2
![]()
![]()
![]()
![]()
![]()
![]() │
║ ║
│
║ ║
CН=CH2 CH─CH2Br CH─CH2─OCCH3
║
O
O O O O
║ ║ ║ ║
CHBr─CH2─OCCH3 2AgOCCH3 CH3CO─CH─CH2─OCCH3 +4H2O
│ │
CHBr─CH2─OCCH3 CH3CO─CH─CH2─OCCH3
║ ║ ║
O O O
2CH2─CH─CH─CH2
![]() │ │ │ │
│ │ │ │
OH OH OH OH
Стереоизомерные эритриты – твёрдые, отлично растворимые в воде, сладкие на вкус вещества.
Пентаэритрит (тетраоксинеопентан) С(СН2ОН)4 в природе не встречается. Это твёрдое высокоплавкое (т. пл. 262°С) вещество. Получается синтетически взаимодействием формальдегида с водным раствором ацетальдегида в щелочной среде:
Ca(OH)2
![]() СН3─С=О + 4НСН=О + Н2О C(CH2OH)4 + H─C─OH
СН3─С=О + 4НСН=О + Н2О C(CH2OH)4 + H─C─OH
│ ║
H пентаэритрит O
муравьиная кислота
Пентиты и гекситы
CH2─CH─CH─ СН─CH2 CH2─CH─CH─ СН─СН─CH2
│ │ │ │ │ │ │ │ │ │ │
OH OH OH OH ОН OH OH OH OH ОН ОН
пентит гексит
Твёрдые, растворимые в воде вещества, сладкие на вкус. Для каждого из спиртов известно много стереоизомеров. Некоторые пентиты и гекситы встречаются в природе, например пентит адонит (в Adonis vernalis), стереоизомерные гекситы – маннит, дульцит, сорбит, идит. Все они имеют нормальный углеродный скелет и могут быть получены восстановлением соответствующих сахаров, которые являются их моноальдегидами.
НЕПРЕДЕЛЬНЫЕ СПИРТЫ
Одноатомные ненасыщенные спирты.
Олефины не могут нести гидроксил при углероде во втором валентном состоянии.
\ \
Структуры С=С─ неустойчивы и изомеризуются в С─С─ (правило Эльтекова ―
/ │ /│ ║
ОН Н О
Эрленмейера). Лишь в некоторых случаях такая изомеризация в заметной степени обратима и мы имеем дело с таутомерным равновесием:
\ \
С=С─ Û С─С─
/ │ /│ ║
ОН Н О
Для структур, в которых не несущий гидроксила непредельный атом не связан с электронооттягивающими группами (─ С─, NO2 и др.), правило Эльтекова-Эрленмейера
║
О
Имеет полную силу. Поэтому виниловый спирт и его гомологи не существуют, а при попытках их получить – перегруппировываются в ацетальдегид (и соответственно его гомологи) или в кетоны:
СН2=СН → СН3─ С─Н
│ ║
ОН О
Причина перегруппировки – проявление того же (мезомерного) эффекта, что и в хлористом виниле, но в этом случае подходящего до конца – до полной передачи электронных пар – и являющегося таким образом +Т-эффектом:
Н Н Н
![]()
![]()
![]() │ ** _ │ │
│ ** _ │ │
![]()
![]() СН2=С─ О─Н → СН2─С=О Н+ → СН3─ С=О
СН2=С─ О─Н → СН2─С=О Н+ → СН3─ С=О
**
Эффект этот протонизирует водород гидроксила и создаёт у второго ненасыщенного атома углерода с его δ- зарядом удобное место атаки для иона водорода. В результате происходит изомеризация – переход протона к углероду.
Однако алкоголяты, а также простые и сложные эфиры винилового спирта не только существуют, но в последних двух случаях даже используются в промышленном масштабе в качестве мономеров. Разумеется, их приходится получать не прямым путём. При действии металлического лития или натрия в растворе в жидком аммиаке на ртутное производное ацетальдегида получаются алкоголяты винилового спирта (И.Ф. Луценко):
ClHgCH2─C=O + 2Me → CH2=C─OMe + MeCl + Hg, где Me = Li или Na.
│ │
H H
Простые и сложные виниловые эфиры получают присоединением к ацетилену спиртов (в присутствии КОН) и карбоновых кислот (в присутствии солей двухвалентной ртути, кадмия, цинка):
KOH
![]() ROH + HC≡CH RO─CH=CH2
ROH + HC≡CH RO─CH=CH2
Me2+; 70°C
![]() R─C─OH + HC≡CH R─C─O─CH=CH2
R─C─OH + HC≡CH R─C─O─CH=CH2
║ ║
O O
![]() Из виниловых эфиров особенно важен винилацетат, полимеризующийся гомолитически в поливинилацетат. Последний используется для получения прозрачных пластмасс, в производстве триплекса (склеивание слоёв силикатного стекла) и для получения поливинилового спирта гидролизом поливинилацетата: nCH3COOCH=CH2
Из виниловых эфиров особенно важен винилацетат, полимеризующийся гомолитически в поливинилацетат. Последний используется для получения прозрачных пластмасс, в производстве триплекса (склеивание слоёв силикатного стекла) и для получения поливинилового спирта гидролизом поливинилацетата: nCH3COOCH=CH2
 H2O
H2O
![]()
 …- ¾СН2─СН─ CН2─СН─ ─… …- ─СН2─СН─СН2─СН─ ─…─
…- ¾СН2─СН─ CН2─СН─ ─… …- ─СН2─СН─СН2─СН─ ─…─
│ │ │ │
СН3С─О CН3С─О ОН ОН
║ ║
O O n /3 n/3
поливинилацетат оливиниловый спирт
Аллиловый спирт СН2=СН─СН2ОН – наиболее простой из непредельных спиртов с удалённым от двойной связи положением гидроксильной группы – по свойствам гидроксила мало отличается от алканолов. Само собой разумеется, что наличие двойной связи обусловливает его непредельные свойства и ряд характерных для непредельных углеводородов реакций. Промышленный способ получения аллилового спирта – гидролиз хлористого аллила, получаемого хлорированием пропилена при высокой температуре:
+OH-
![]()
![]() CH2=CH─CH3 + Cl2 CH2=CH─CH2Cl CH2=CH─CH2OH
CH2=CH─CH3 + Cl2 CH2=CH─CH2Cl CH2=CH─CH2OH
-HCl
Ацетиленовые спирты
Эти вещества не получили большого значения и изучены сравнительно мало. Назовём из них один пропаргилловый спирт СН≡С─СН2ОН, который в настоящее время проще всего получают по методу Реппе:
CuC≡CCu
![]() НС≡СН + СН2О СН≡С─СН2ОН
НС≡СН + СН2О СН≡С─СН2ОН
Ацетилен
Он обладает обычной спиртовой функцией, при замене гидроксила способен к аллильной перегруппировке; имея ацетиленовый водород, может замещать его, как и ацетилен, на металлы, в частности на серебро и медь.
Бутиндиол НОСН2─С≡С─СН2ОН используется при получении бутадиена-1,3:
H3PO4
![]() НОСН2─С≡С─СН2ОН
Н2С——СН2
НОСН2─С≡С─СН2ОН
Н2С——СН2
- H2O │ │
H2C CH2
\ /
O
NaPO3
![]() Н2С——СН2
CH2=CH─CH=CH2
Н2С——СН2
CH2=CH─CH=CH2
│ │ - H2O
H2C CH2
\ /
O
АРОМАТИЧЕСКИЕ ОКСИСОЕДИНЕНИЯ
ФЕНОЛЫ
Термин «фенолы» происходит от старинного названия бензола «фен», введённого Лораном (1837 г.), и обозначает ароматическое вещество, содержащее гидроксил, связанное непосредственно с углеродом ароматического ядра. Фенолы, как и спирты могут содержать в своём составе, как одну, так и несколько гидроксильных групп. В зависимости от чиисла гидроксильных групп в молекуле различают одно-, двух-, трёх- и многоатомные фенолы.
Структура и номенклатура.
Фенолы обычно называют как производные простейшего члена этого ряда - фенола. Для метилфенолов имеется специальное название - крезолы.
OH OH OH OH OH OH
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Cl OH гидрохинон
Cl OH гидрохинон
CH3 OH
Фенол о-хлорфенол м-крезол пирокатехин резорцин OH
OH OH
![]()
![]()
![]()
![]() Br Br Cl
Br Br Cl
Br NO2 2,4,6 - трибромфенол 2-хлор-4-нитрофенол
Физические свойства.
Табл. Фенолы
| Фенол | Т. плавления, °С | Т. кип., °С | Плотность, г/см3 |
| Фенол Крезол о-, или 1,2- м-, или 1,3- п-, или 1,4- | 41 30 11 36 | 182 191,5 202,8 202,5 | 1,072 1,0465 1,034 1,035 |
Простейшие фенолы представляют собой жидкости или низкоплавкие твёрдые вещества; из-за образования водородных связей они обычно имеют высокие температуры кипения. Сам фенол заметно растворим в воде (9г. на 100г. воды), из-за оразования водородных связей с водой; большинство других фенолов практически не растворимы в воде. Фенолы - бесцветные вещества, если только они не содержат каких либо групп, обусловливающих появление окраски.
Простейший из фенолов – оксибензол (собственно, фенол) и его гомологи: о-, м- и п-крезолы содержатся в каменноугольной смоле. Дополнительные количества фенола, мировое потребление которого достигает миллионов тонн, получаются из бензола. Для этого используется (всё в меньших масштабах) старый метод щелочного плавления соли бензолсульфокислоты: 300 °C
![]() C6H5SO3Na + Na OH C6H5OH + Na2SO3
C6H5SO3Na + Na OH C6H5OH + Na2SO3
Некоторое количество фенола получают гидролизом хлорбензола перегретым паром (450-500°С) над катализатором – силикагелем, промотированным ионами Cu2+ (Рашиг):
Силикагель: Cu2+
![]() C6H5Cl +H2O C6H5OH +HCl
C6H5Cl +H2O C6H5OH +HCl
Наибольшие перспективы развития имеет разложение перекиси кумола (изопропилбензола) разбавленными кислотами. Процесс состоит в следующем:
ООН
│
СН3─ СН─СН3 СН3─ С─СН3
![]()
![]() Н+ │ О2
│
Н+ │ О2
│
![]()
![]()
![]()
![]() + СН3─СН=СН2 → → →
+ СН3─СН=СН2 → → →
ОН
│
![]()
→ + СН3─ С─СН3
║
О
Фенол – слабая кислота с константой диссоциации при комнатной температуре в водном растворе 1,3∙10-10.
Таким образом, он на несколько порядков кислее воды, не говоря уже о жирных спиртах, но гораздо слабее уксусной кислоты (1,8∙10-5). Фенол умеренно растворим в воде (8% при 15°С). Вода растворяется в феноле с образованием жидкого при комнатной температуре раствора. Сам фенол – бесцветное легкоплавкое (+41°С) кристаллическое вещество, вследствие окисления розовеющее на воздухе. Крезолы менее, чем фенол растворимы в воде, подобно фенолу хорошо растворимы в эфире, спиртах, хлороформе, бензоле.
Фенолы хорошо растворяются в водных растворах щелочей в результате образования фенолятов щелочных металлов:
![]()
![]() ArOH + NaOH ArO- Na+ + H2O
ArOH + NaOH ArO- Na+ + H2O
Гидролиз фенолята (обратная реакция) вследствие слабости кислотных свойств фенола заходит далеко, и требуется избыток щёлочи, чтобы сместить равновесие вправо. Уже двуокись углерода выделяет фенол из раствора фенолята.
Кислотные свойства фенольного гидроксила вызваны мезомерным взаимодействием с ароматическим ядром, что выражается символами:
H
![]() ½ H+ H+ H+
½ H+ H+ H+
O O O O
![]()
![]()
![]()
![]()
![]() ½ ║ * ║ * ║
½ ║ * ║ * ║
![]()
![]()
![]()
![]()
![]()
![]()
![]() * *
* *
* *
Валентные электроны атома кислорода (в том числе и связывающие водород с кислородом) оказываются частично рассредоточенными в орто- и пара-положения бензольного ядра, а водородный атом гидроксила – протонизированным. Таким образом, бόльшая кислотность фенола (сравнительно со спиртами) – это другая сторона сильного орто-пара-ориентирующего действия гидроксила в реакциях электрофильного замещения
Реакции гидроксила фенолов
1. Образование фенолятов (см. выше).
2. Образование простых эфиров фенолов алкилированием фенолятов:
ArONa + RI → ArOR + NaI
ArONa + (CH3O)2SO2 → ArOCH3 + CH3O─SO2ONa
3. Образование сложных эфиров фенолов (в отличие от сложных эфиров спиртов) не может быть достигнуто взаимодействием их с кислотами, а только ацилированием фенолов (лучше в щелочной среде) галоидангидридами или ангидридами кислот:
![]() ArONa + Cl─ C─R ArO─ C─R + NaCl
ArONa + Cl─ C─R ArO─ C─R + NaCl
║ ║
O O
O=C─R
│
![]() ArONa +
O ArO─C─R + R─ C─ONa
ArONa +
O ArO─C─R + R─ C─ONa
│ ║ ║
O=C─R O O
4. Замещение гидроксила на хлор при действии PCl5 протекает гораздо труднее, чем для спиртов, и с плохим выходом. В этом случае происходит главным образом хлорирование в ядро, причём PCl5 превращается в PCl3. С PCl3 в малой степени идёт замещение гидроксила на хлор, а в большей степени – образование трифенилфосфита (эфира фосфористой кислоты). С хлорокисью фосфора POCl3 образуется фениловый эфир фосфорной кислоты.
5. При перегонке с цинковой пылью фенолы превращаются в углеводороды:
ArOH + Zn → ArH + ZnO
Реакции ароматического ядра фенолов
Гидроксил – один из сильнейших, а в щелочном растворе сильнейший орто-пара-ориентант. В соответствии с этим для фенолов легко проходят реакции электрофильного замещения.
Механизм электрофильного замещения в фенолах обычно отличается от замещения в бензоле, его гомологах и даже в эфирах фенолов. Это отличие связано с лёгкостью гетеролиза связи О─Н, поскольку вместо нестабильного и заряженного σ-комплекса промежуточно получается сравнительно устойчивое соединение с хиноидной структурой типа I:
![]()
![]() O¾H O O¾H
O¾H O O¾H
![]()
![]() ½
(1) ║ (2) ½
½
(1) ║ (2) ½
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() + A+
+ A+
![]()
![]()
![]()
/\ ½
H A A
I
При этом установлено, что для большинства реакций фенолов первая стадия – быстрая и обычно обратимая, а вторая – медленная. В ряде случаев соединения типа I были выделены в свободном виде, правда, только для тех фенолов, в которых заняты все орто- и пара-положения (в случае обычных фенолов ароматизация совершается слишком быстро). Например:
OH O
![]()
![]()
![]() Br ½ Br Br
║ Br
Br ½ Br Br
║ Br
![]()
![]()
![]()
![]()
![]()
![]() HNO3
HNO3
![]()
![]()
![]() ½
H3C NO2
½
H3C NO2
Br
Если в феноле о- и п-положения заняты, то может происходить (особенно при нитровании) замена имеющихся заместителей на другие группы. Лёгкость такого замещения увеличивается в следующей последовательности: Br<SO3H<H. Замена карбоксильной группы происходит даже при азосочетании.
Галогенирование фенолов.
В неводной среде галогенирование фенолов при соответствующих соотношениях реагентов приводит к смеси о- и п-галогенфенолов, далее к 2,4-дигалогенфенолам и, наконец, к 2,4,6-тригалогенфенолам (их лучше получать в водной щелочной среде). В случае орто- и пара-замещённых фенолов, например крезолов, занятые заместителем (например, метилом) места галогенированием не затрагиваются.
Бромирование фенола избытком бромной воды проходит по схеме:
OH OH O
![]()
![]()
![]()
![]()
![]()
![]()
![]() ½ Br ½ Br Br ║ Br
½ Br ½ Br Br ║ Br
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() +3Br2 +Br2
+3Br2 +Br2
![]()
![]() -3HBr -HBr
-3HBr -HBr
Br Br Br
Ориентирующая сила гидроксила, т.е. сообщение гидроксилом нуклеофильной активности п-углеродному атому, такова, что этот углерод и после замещения связанного с ним водородного атома способен воспринять электрофильную атаку электроположительного атома брома. Присоединение второго атома брома закрепляет циклогексадиеновую структуру.
![]()
![]() O¾H O
O¾H O
![]()
![]()
![]()
![]()
![]()
![]() Br ½ Br Br ║ Br
Br ½ Br Br ║ Br
Br Br-Br Br Br + Br-+ H+
Сульфирование фенолов.
Сульфирование фенола при комнатной температуре даёт в основном о-фенолсульфокислоту, при 100°С получается п-изомер, а в более жёстких условиях – 2,4-фенолдисульфокислота.
Нитрование фенолов.
Для получения мононитрофенолов приходится нитровать фенолы на холоду разбавленной азотной кислотой (~30%-ной), лучше всего получаемой смешением водного раствора селитры с серной кислотой (чтобы избежать присутствия окислов азота). Образуется смесь о- и п-нитрофенолов, из которой о-нитрофенол удаляют отгонкой с водяным паром, а п-изомер выделяют кристаллизацией. м-Изомер приходится готовить обходным путём, например из м-нитроанилина через м-нитрофенилдиазоний. 2,4-Динитрофенол проще всего получить гидролизом 2,4-динитрохлорбензола.
Тринитрофенол, называемый пикриновой кислотой, производят в промышленном масштабе, нитруя крепкой нитрующей смесью 2,4-фенолдисульфокислоту, получаемую сульфированием фенола, без выделения её из сульфирующей массы. При этом нитруется не только свободное шестое положение, но и сульфогруппы замещаются на нитрогруппы. Наличие в феноле сульфогрупп защищает его и от окисления и от действия окислов азота.
Нитрозирование фенолов.
При действии водного раствора азотистой кислоты фенол нитрозируется в пара-положение:
НО─ + HO─N=O → HO─ ─N=O
Нитрозофенол таутомерен монооксиму п-бензохинона:
HO─ ─N=O ↔ O= =N─OН
![]()
Электрофильные замещения в фенолах с образованием углерод-углеродной связи.
Таких реакций известно много. Они используются для получения бифункциональных соединений, например фенолокислот, фенолоальдегидов и фенолоспиртов.
При нагревании фенолята натрия в токе СО2 образуется салициловокислый натрий (реакция Кольбе):
![]()
![]() ONa OH O
ONa OH O
![]()
![]() │
│ C
│
│ C
![]()
![]()
![]()
![]() ONa
ONa
+ CO2 →
При действии на фенолят натрия (избыток щёлочи) четырёххлористого углерода также образуется салициловокислый натрий, а при действии хлороформа – салициловый альдегид:
![]() ONa
OH ONa
ONa
OH ONa
![]()
![]()
![]() ½ NaOH ½ C
½ NaOH ½ C
![]()
![]()
![]()
![]()
![]() + CCl4 O + NaCl + H2O
+ CCl4 O + NaCl + H2O
![]() ONa
OH H
ONa
OH H
![]()
![]()
![]()
![]()
![]() ½ NaOH ½ C
½ NaOH ½ C
![]()
![]()
![]()
![]() + CHCl3 O + NaCl + H2O
+ CHCl3 O + NaCl + H2O
Действием олефинов на фенолы в присутствии льюисовых кислот получают п-алкилфенолы (частный случай реакции Фриделя-Крафтса):
ОН OH
│ ZnCl2 │
![]()
![]()
![]()
![]() + RCH=CH2
+ RCH=CH2
│
RCH─CH3
C синильной кислотой (или нитрилами) в присутствии хлористого водорода фенолы дают иминоальдегидофенолы или иминокетонофенолы (реакция Геша), а после гидролиза иминогруппы получаются сами оксиоксосоединения:
OH OH OH
│ HCl │ H2O (H+) │
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() + XCN
+ XCN
│ │
X─C=NH X─C=O
(X=H, арил или алкил)
Наиболее важная реакция этого рода – реакция фенолов с формальдегидом, которая протекает в присутствии как кислот, так и щелочей. При нагревании фенола (избытка) с формалином и серной кислотой происходит бурная реакция и образуется растворимый в спиртах, ацетоне и сложных эфирах полимер линейного строения – «новолак». При щелочной конденсации фенола с избытком формалина сначала образуется легкоплавкий сравнительно низкомолекулярный полимер «резол», подобно новолаку растворимый в органических растворителях. Это – так называемый термореактивный полимер: при нагревании происходит дальнейшая конденсация свободных оксиметиленовых групп с образованием метиленовых мостов, и полимер приобретает сетчатую структуру. Получаемый «резитол» нерастворим в органических растворителях, но сохраняет некоторую пластичность. При нагревании до 150°С конденсация идёт дальше и получается химически очень устойчивый, неплавкий и нерастворимый полимер – «резит», который можно нагревать до температуры ~300°С. Таковы три стадии процесса конденсации, объединяемые названием «бакелитизация» (по имени изобретателя бакелита – Бакеланда). Обычно резол перед последующей стадии конденсации смешивают с наполнителем (минеральным типа асбеста или органическим типа древесины, лигнина, целлюлозы) или пропитывают им древесину или волокнистые материалы и затем подвергают дальнейшей бакелитизации. Этот открытый в 1909 г. тип феноло-формальдегидных пластмасс и в настоящее время сохранил своё значение.
Химический смысл протекающих процессов выражается следующей примерной схемой:
OH
![]()
![]() ½ CH2OH
½ CH2OH
OH OH OH
![]()
![]()
![]() ½ ½ ½
½ ½ ½
![]()
![]()
![]()
![]()
![]()
![]() + CH2O OH
CH2
+ CH2O OH
CH2
![]() ½
½
![]()
![]()
![]()
![]()
![]()
![]()
![]() ½
½
![]()
![]()
![]() CH2OH
CH2OH
![]()
![]() ½
½
CH2OH

OH OH OH
![]()
![]()
![]()
![]() ½ ½ ½
½ ½ ½
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() CH2 CH2
CH2 CH2
![]() OH
OH
![]() OH
OH OH OH
OH
OH OH OH
![]()
![]()
![]()
![]()
![]()
![]() ½ ½ ½
½ ½ ½
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() CH2 …¾CH2
CH2 …
CH2 …¾CH2
CH2 …
½ ½
![]() CH2OH CH2
CH2OH CH2
OH ½
![]()
![]()
![]() OH ½ OH
OH ½ OH
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() ½
… CH2
½
… CH2
![]()
![]()
![]()
![]() CH2
OH ½
CH2
OH ½
![]() ½
CH2
½
CH2
CH2 ½
![]() ½
½
![]()
Таким образом происходит постепенное «сшивание» метиленовыми мостами всё большего количества молекул фенола в хаотически построенные макромолекулы резола, резитола и, наконец, резита. Химическая стой кость резита объясняется не только тем, что значительное количество активных орто- и пара-положений фенола замещены метиленовыми группами, сколько тем, что в следствие полной нерастворимости бакелита реагенты могут действовать на него только с поверхности.
Алифатические кетоны в кислой среде реагируют с фенолом, образуя ди-n-оксифенилоктаны:
CH3
![]()
![]() (H+) ½
(H+) ½
![]()
![]()
![]() CH3COCH3 + 2C6H5OH HO¾ ¾C¾ ¾OH + H2O
CH3COCH3 + 2C6H5OH HO¾ ¾C¾ ¾OH + H2O
½
CH3
Такой 2,2-бис-(4'-оксифенил)-пропан (т.н. дифенилолпропан) применяется в синтезе пластмасс повышенной теплостойкости, получаемых путём этерификации фенольных гидроксилов ароматическими двухосновными кислотами типа терефталевой.
МНОГОАТОМНЫЕ ФЕНОЛЫ
Диоксибензолы
Изомерные диоксибензолы носят следующие названия: о-диоксибензол – пирокатехин, м-изомер – резорцин и п-изомер – гидрохинон. Это хорошо растворимые в воде, твёрдые, лишённые запаха вещества.
Пирокатехин известен как продукт декарбоксилирования при нагревании пиротокатеховой кислоты, находимой в растениях:
![]()
![]() t °C
t °C
![]()
![]() НО─ ─С─ОН → НО─
+ СО2
НО─ ─С─ОН → НО─
+ СО2
![]()
![]() ║
║
HO O HO
Пирокатехин – сильный восстановитель, и, окисляясь гетеролитически (например, ионом Ag+), он превращается в о-бензохинон:
![]()
![]() OH O
OH O
![]()
![]()
![]()
![]()
![]() [O]
[O]
![]()
![]()
![]()
![]()
![]() + H2O
+ H2O
OH O
Резорцин (м-оксибензол) получают в технике сплавлением со щёлочью м-бензолдисульфоната натрия:
![]()
![]()
![]()
![]()
![]() SO3Na ONa
SO3Na ONa
![]()
![]() + NaOH + 2Na2SO3
+ NaOH + 2Na2SO3
![]()
![]()
SO3Na ONa
Резорцин устойчивее своих изомеров к окислению. Кислотные его свойства выражены сильнее, чем у фенола. Уже водородом в момент выделения (амальгама натрия и вода) он восстанавливается в дигидрорезорцин (циклогександион-1,3):
![]()
![]() OH O
OH O
![]() ½ H2C CH2
½ H2C CH2
![]()
![]()
![]()
![]() + 2H
+ 2H
![]()
![]() H2C O
H2C O
OH CH2
Резорцин ещё легче, чем фенол, воспринимает разнообразные электрофильные атаки, так как обе его гидроксильные группы осуществляют согласованную ориентацию. Поэтому резорцин легко галоидируется, сульфируется, нитруется, нитрозируется и пр. Одно из его главных применений – синтез азокрасителей, в котором он служит азосоставляющей.
При исчерпывающем нитровании резоцина получается тринитрорезорцин, стифниновая кислота:
OH
![]()
![]()
![]() O2N
NO2
O2N
NO2
![]()
½ OH
NO2
во многом напоминающая пикриновую кислоту. Для карбоксилирования резоцина достаточно нагреть его в растворе бикарбоната натрия:
ONa OH
![]()
![]() ½ ½
½ ½
![]()
![]()
![]() + CO2
+ CO2
![]()
![]() ONa OH
ONa OH
½
O=C¾OH
Получаемое соединение носит название резоциловой кислоты.
Гидрохинон получают восстановлением п-бензохинона:
![]()
![]() О= =О + 2Н НО─ ─ОН
О= =О + 2Н НО─ ─ОН
![]()
Как и пирокатехин, гидрохинон – сильный восстановитель, при окислении образующий п-бензохинон.
Пирокатехин и гидрохинон применяются как фотографические проявители, восстанавливающие бромистое серебро до металла.
Полиоксибензолы
Смежный триоксибензол называется пирогаллолом, так как получается пиролизом (декарбоксилированием) галловой кислоты:
![]()
![]() HO HO
HO HO
![]()
![]()
![]()
![]()
![]() HO¾ ¾C¾OH HO¾ + CO2
HO¾ ¾C¾OH HO¾ + CO2
![]()
![]() ║
║
HO O HO
выделяемой из продуктов гидролиза дубильных веществ типа танина.
Пирогаллол в щелочных растворах легко окисляется даже кислородом воздуха, поэтому такие растворы используются для поглощения кислорода. В фотографии пирогаллол применяется как проявитель.
Симметрический триоксибензол – флороглюцин в виде его производных очень распространён в растительном мире.
Обычно флороглюцин получают гидролизом симметрического триаминобензола (его готовят восстановлением тринитробензола):
![]()
![]() H2N HO
H2N HO
![]()
![]() H+
H+
![]()
![]()
![]() ¾NH2 + 3H2O
¾OH + 3NH+
¾NH2 + 3H2O
¾OH + 3NH+
H2N HO
По свойствам флороглюцин похож на резорцин.
1,2,4-Триоксибензол можно синтезировать, присоединяя к п-бензохинону уксусный ангидрид и гидролизуя образовавшийся ацетет.
Гексаоксибензол получают подкислением продукта соединения металлического калия и окиси углерода:
OK OH
![]()
![]()
![]()
![]() KO ½ OK HO ½ OH
KO ½ OK HO ½ OH
![]()
![]()
![]()
![]() H+
H+
![]()
![]()
![]()
![]()
![]()
![]() 6СО + 6К +
6K+
6СО + 6К +
6K+
KO ½ OK HO ½ OH
OK OH
Похожие работы


... синтетическим красителям и цели работы. Новые материалы требуют новых красителей, а потому тема по синтезу красителей всегда остаётся актуальной. Цель данной работы – синтез 4-окси-3-карбоксиазобензола, который является представителем большого класса азокрасителей. На примере его получения изучить условия проведения реакций диазотирования 4-окси-3-карбоксиазобензола и реакции. 2. Современная ...
... , причем преобладают кислые. Количество отдельных групп аминокислот в белках зависит от зоотехнических факторов, что и обуславливает их физико-химический состав. Молоко по содержанию незаменимых аминокислот является полноценным. Состав незаменимых АК в некоторых белках % Аминокислоты Идеальный белок Казеин Сывороточные белки молока Белок яйца Белок пшеницы Белок ...
... витамина А - с 22 углеродными атомами и 6 конъюгированными двойными связями. Наличие этого гомолога названного витамином А2, отмечено в жирах пресноводных рыб. БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ КАРОТИНА И КАРОТИНОИДОВ Биологическая активность каротина, т.е. степень способности его оказывать на организм такое же действие, как и витамин А, зависит эффективность процесса его усвоения и ...
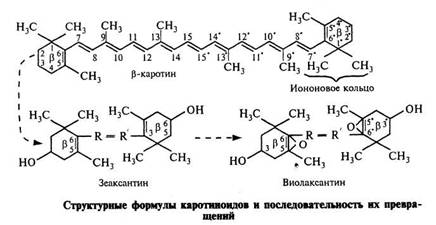
... ксантофилл зеаксантин. В независимой от света реакции благодаря включению кислорода происходит обратное превращение зеаксантина в виолаксантин. Возможно, этот цикл служит для удаления излишков кислорода, образующихся при фотолизе воды. В верхушках побегов растений каротиноиды обеспечивают определение направления света и их ориентацию к световому потоку за счет фототропизма. 1.4 Карот
0 комментариев