Навигация
Иод в-во летучее, поэтому титрование ведут на холоду. При нагревании чувствительность крахмала уменьшается
64431
знак
7
таблиц
0
изображений
1. иод в-во летучее, поэтому титрование ведут на холоду. При нагревании чувствительность крахмала уменьшается.
2. йодометрическое титрование нельзя проводить в щелочной среде, т.к. I2взаимодействует со щелочами. I2+2NaOH↔NaI+NaIO+H2O. Гипоиодид ион IO- является более сильным окислителем, чем I2 и может окислить тиосульфат до сульфат ионов: S2O32-→SO42-.
3. растворимость иода в воде мала, поэтому при измерении окислителей необходимо применять избыток КI. Это способствует растворению выделенного из реакции иода: КI+I2→K[I3].
4. реакция между иодид ионом и окислителем идёт не достаточно быстро, поэтому к титрованию приступают через некоторое время после прибавления ок-ля (р-р оставляют на 5 мин. до завершения реакции).
5. перед началом титрования реакционную смесь оставлять в тёмном месте, т.к. свет ускоряет в кислых р-рах побочную реакцию – окисление иодид ионов до иода кислородом воздуха: 2Na2S2O3+O2→2Na2SO4+2S↓
Прямое титрование. Это измерение восст-ля. Проводя прямое титрование необходимо помнить о летучести иода. Ст. р-ром в прямом титровании является р-р иода, а индикатором крахмал, который добавляют сразу. Титруют до появления светло синей окраски.
Химизм. 2Na2S2O3+I2→2NaI+Na2S4O6 – тетротионат натрия.
Расчёт: m(Na2S2O3)=V(I2)*C(1/zI2)*M(1/z Na2S2O3)
При прямом титровании возможны погрешности, а именно: 1.∆Е – мала, реакция идёт медленно особенно в конце титрования и не успевший прореагировать иод окрашивает крахмал, что приводит к заниженным результатам анализа.2.Восст-ль может окисляться кислородом воздуха, получается заниженный результат.
10.Измерение окислителей йодометрическим методом (К2Сr2О7, КВrО3). Написать уравнения реакций и расчётные формулы.
В-во бромата калия является ок-ем, поэтому его измеряют методом титрования заместителя. К контр. р-ру добавляют р-р H2SO4 c C(1/z)=2моль/л, V=0,001-0,010л, р-р KI с ω%=10% и V=0,005л и поставить в тёмное место на 5 мин. для завершения реакции. Титровать Na2S2O3 без индикатора. Когда окркска титруемого р-ра станет бледно-жёлтой прибавить р-р крахмала 1-2 кап. и титровать до исчезновения синей окраски.
Химизм. KBrO3+KI+H2SO4→KBr+I2+K2SO4+H2O
I2+Na2S2O3→NaI+Na2S4O6
Расчёт. M(KBrO3)=V(Na2S2O3)*C(1/z Na2S2O3)*M(1/zKBrO3)
11.Измерение окислителей (Cu в CuSO4*5H2O) и хлороводородной к-ты йодометрическим методом. Написать уравнения реакций и расчётные формулы.
Методом титрования заместителя можно определить медь в медном купоросе. Ст. р-ром титруют не измеряемый компонент, а его заместитель, который выделяется (в эквивалентном кол-ве) при реакции измеряемого компонента с другим специально добавленным реактивом.
Химизм. 2CuSO4+4KI→2CuI↓+I2+2K2SO4, I2 – заместитель
I2+2Na2S2O3→2NaI+Na2S4O6
Расчёт: m(Cu)=V(Na2S2O3)*C(1/z Na2S2O3)*M(1/zCu), Z=1, Cu2++1e→Cu.
Измерение хлороводородной к-ты.
Химизм. КIO3+5KI+6HCl→3I2+6KCl+3H2O
I2+2Na2S2O3→2NaI+Na2S4O6.
Расчёт. M(HCl)=V(Na2S2O3)*C(1/z Na2S2O3)*M(1/zHCl).
12.Виды титрования в йодометрии. Привести примеры, химизм реакций, расчётные формулы.
Существует три вида титрования в йодометрии: прямое, обратное и замещение.
Прямое титрование. Это измерение восст-ля. Проводя прямое титрование необходимо помнить о летучести иода. Ст. р-ром в прямом титровании является р-р иода, а индикатором крахмал, который добавляют сразу. Титруют до появления светло синей окраски.
Химизм. 2Na2S2O3+I2→2NaI+Na2S4O6 – тетротионат натрия.
Расчёт: m(Na2S2O3)=V(I2)*C(1/zI2)*M(1/z Na2S2O3)
При прямом титровании возможны погрешности, а именно: 1.∆Е – мала, реакция идёт медленно особенно в конце титрования и не успевший прореагировать иод окрашивает крахмал, что приводит к заниженным результатам анализа.2.Восст-ль может окисляться кислородом воздуха, получается заниженный результат.
Обратное титрование. Лучшие результаты получаются методом обратного титрования. Анализируемый р-р обрабатывают избыточным кол-вом точно отмеренного р-ра иода. А его избыток оттитровывают р-ром тиосульфата. Начинают титровать без индикатора (крахмал не доб.) титруют до бледно жёлтого окрашивания р-ра. Затем добавляют крахмал и титруют до исчезновения синей окраски. Крахмал добавляют тогда, когда иода в р-ре останется мало (бледно-жёлтый р-р). Крахмал адсорбирует иод и адсорбированный иод медленно реагирует с Na2SO3, что приводит к погрешности анализа – завышенный результат.
Химизм: I2+Na2S2O3→2NaI+Na2S4O6 ; Na2SO3+I2+H2O→Na2SO4+2HI
Расчёт: m(Na2SO3)=[С(1/zI2)*V(I2)-C(1/z Na2S2O3)*V(Na2S2O3)]* M (1/z Na2SO3)
Метод замещения. Методом титрования заместителя можно определить медь в медном купоросе. Ст. р-ром титруют не измеряемый компонент, а его заместитель, который выделяется (в эквивалентном кол-ве) при реакции измеряемого компонента с другим специально добавленным реактивом.
Химизм. 2CuSO4+4KI→2CuI↓+I2+2K2SO4, I2 – заместитель
I2+2Na2S2O3→2NaI+Na2S4O6
Расчёт: m(Cu)=V(Na2S2O3)*C(1/z Na2S2O3)*M(1/zCu), Z=1, Cu2++1e→Cu.
13.Пермангонатометрический метод анализа. Общая характеристика метода. Ст. р-ры, их приготовление.
Метод пермангонатометрии основан на реакциях окисления восст-ей ионом перманганата. Окисление может проводиться как в кислой, так и в щелочной или нейтральной среде.
При окислении в кислой среде марганец (VII) в составе КМnО4, применяемого для окисления, восст-ся до Мn2+. MnO4-+8H++5e→Mn2++4H2O, Е0=1,51В.
При окислении в щелочной или нейтральной среде марганец (VII) восст-ся до марганца (IV), причём образует двуокись марганца:
MnO4-+2H2O+3e→MnO2↓+4OH-, Е0=0,6В – в нейтральной
MnO4-+1е→MnO42-, Е0=0,56В – в щелочной.
Стандартный потенциал пары MnO4-/ Mn2+ выше, чем в I и II случаях, следовательно, окислительная способность перманганата в кислой среде выше, чем в щелочной перманганатометрические измерения проводят в кислой среде, т.к. точку эквивалентности установить легко по обесцвечиванию р-ра перманганата калия. Образование же MnO2 затрудняет установление точки эквивалентности, т.к. выпадает в тёмно-бурый осадок.
Приготовление стандартного р-ра KMnO4. титрование проводят без индикатора, реакция чувствительна. Одна капля КМnО4 с С(1/z)=0,01моль/л окрашивает в конце титрования 50мл р-ра в отчётливо розовый цвет. КМnО4 не отвечает требованиям к исходным в-вам , т.к. содержит примеси продуктов восст-ия MnO2. легко разлагается под влиянием восст-ей: NH3, органические в-ва, попадающих в воду с пылью. Поэтому КМnО4 готовят из приблизительной навески, а затем устанавливают точную конц-ию по другому ст. р-ру. Чтобы приготовить р-р нужно рассчитать навеску:
m(КМnО4)=V(р-ра)*С(1/z КМnО4)*M(1/z КМnО4).
Навеску берём на тех. весах. Воду отмеряем цилиндром, растворение ведём в горячей воде, т.к. КМnО4 растворяется медленно. Приготовленный охлаждённый р-р переливают в тёмную посуду. Закрывают стеклянной пробкой и оставляют на 7-10 дней для выстаивания в темноте (за это время окислятся все случайные органические примеси в воде, а образовавшийся MnO2 сядет на дно). Р-р сливают или фильтруют через стеклянную вату, или через стеклянный фильтровачный тигель с пористым дном.
14.Перманганатометрический метод анализа. Установка точной конц-ии ст. р-ра в-ва перманганата калия. Написать химизм процесса, расчётные формулы.
Метод пермангонатометрии основан на реакциях окисления восст-ей ионом перманганата. Окисление может проводиться как в кислой, так и в щелочной или нейтральной среде.
Химизм. 5H2C2O4+2KMnO4+3H2SO4→10CO2+2MnSO4+K2SO4+8H2O.
Расчёт. С(1/zKMnO4)=V(H2C2O4*2H2O)*C(1/z H2C2O4)*2H2O/V(KMnO4).
15.Изметение массы общего железа перманганатометрическим методом. Защитная смесь Рейнгарда – Циммермана, её состав и значение.
Для определения железа берут аликвоту, добавляют воды дист., приливают HCl (1:1), бросают несколько капсул Zn и нагревают до обесцвечивания р-ра. Zn отфильтровывают, приливают 10мл защитной смеси Рейндгарда–Циммермана и титруют р-ром KMnO4 с C(1/zKMnO4)=0.1 моль/л до появления не исчезающего в течении 30 сек. бледно-розового окрашивания.
Химизм. Fe3++Zn→Fe2++Zn2+
Fe2++MnO4-+H+→Fe3++Mn2++4H2
FeSO4+KMnO4+H2SO4→Mn2++Fe3+
MnO4-+8H++5e→Mn2++4H2O
2Fe2+-2e→2Fe3+
2MnO4-+16H++10Fe2+→2Mn2++8H2O+10Fe3+
2K+ 8SO42- 10SO42- 2K+ 8SO42- 10SO42-
2KMnO4+8H2SO4+10FeSO4→5Fe2(SO4)3+2MnSO4+8H2O+K2SO4.
Расчёт. ω%(Fe2O3)=C(1/zKMnO4)*V(KMnO4)*M(1/z Fe2O3)*Vкол*100%/m(нав)*Vал
ω%(Fe2O3)=ТKMnO4/Fe2O3*V(KMnO4)*Vкол*100%/m(нав)*Vал.
M(Fe)=C(1/z KMnO4)*V(KMnO4)*M(1/Fe).
В состав смеси Рейндгарда-Циммермана входят H2SO4+H3PO4+MnSO4. Серная-для серы, фосфорная-для связывания Fe в бесцветный комплекс, MnSO4-для уменьшения потенциала перманганата.
ЕMnO4-/Mn2+=E0MnO4-/Mn2++0.059/n(1)*lg*[Fe3+]/[Fe2+].
Перманганат не тратится на Cl- оины, для этого уменьшаем.
16.Виды титрования в перманганатометрии. Привести примеры, химизм реакций, расчётные формулы.
17.Измерение массовой доли MnO2 в пиролюзите перманганатометрическим методом анализа. Написать химизм процесса, расчётную формулу.
Титрование по остатку. Два ст. р-ра: KMnO4 и р-р восст-ля, имеющий потенциал меньше, чем у определяемого ок-ля.
К измеряемому ок-лю добавляют из бюретки избыточный, но точно отмеренный объём рабочего р-ра восст-ля. Избыток восст-ля оттитровываем р-ром KMnO4.
Химизм. MnO2+H2C2O4+H2SO4→MnSO4+2CO2+2H2O
H2C2O4+2KMnO4+3H2SO4→10CO2+2MnSO4+K2SO4+8H2O/
Расчёт.ω%(MnO2)=[V(H2C2O4)*C(1\zH2C2O4)-V(KMnO4)*C(1/zKMnO4)]M(1/zMnO2)*100%/m(нав)
M(MnO2)=[V(H2C2O4)*C(1\zH2C2O4)- V(KMnO4)*C(1/zKMnO4)]M(1/zMnO2).
18.Хроматометрический метод анализа. Сущность, ст. р-ры. Индикация точки эквивалентности. Защитная смесь Кнопа, её состав, назначение компонентов.
В основе метода лежит ОВР. Cr2O7+14H+→2Cr3++7H2O (восст-ие).
Е0Cr2O7/2Cr3+=1.33В.
K2Cr2O7 сильный окислитель, с его помощью измерить все в-ва, у которых стандартный электродный потенциал меньше 1,33В. Для проведения анализа необходима кислая среда.
Достоинства метода. 1. K2Cr2O7 отличается высокой устойчивостью. Р-р готовят из точной навески из химически чистого K2Cr2O7.
m(K2Cr2O7)=V(р-ра)*C(1/z K2Cr2O7)*M(1/z K2Cr2O7). 2. Можно титровать ионы Fe в присутствии хлорид ионов, т.к. их ЭВП примерно равны Е0Cr2/2Cr-=1.35В.
Недостаток. При титровании р-ром K2Cr2O7 образуются ионы зелёного цвета – Cr3+ -, что мешает индикации точки эквивалентности.
Индикаторы. Для фиксирования точки экв-ти применяются редокс-индикаторы. Ок-ая и восст-ая формы индикатора имеют разные окраски. Наиболее часто применяют: дифениламин, дифениламиносульфанат натрия, фенилантрониловая к-та. Е0диф-на=0,76В.
При титровании K2Cr2O7 окраска дифениламина изменяется от бесцветной до фиолетовой (восст-ая ф. - бесцветная, ок-ая ф. - фиолетовая). Если потенциал измеряемых ионов и индикатора близки между собой, то и реакции их пойдут почти одновременно. Изменение окраски индикатора может произойти задолго до точки экв-ти, чтобы этого не произошло надо понизить о-в потенциал пары Fe3+/Fe2+. Для этого надо уменьшить конц-ию ионов Fe3+, связа его в бесцветный комплекс фосфорной кислотой. Н3[Fe(PO4)2].
Смесь Кнопа. В состав входят Н2SO4+H3PO4. Серная – для серы, фосфорная – уменьшить потенциал Fe, чтобы реакция пошла вперёд; используют с индикатором дифениламин или дифениламинсульфонат натрия.∆Ер=Еок-ля-Евосст-ля
∆Ер1=E0K2Cr2O7/2Cr3+-E0Fe3+/Fe2+. ∆Ер2=Е0Cr2O72-/2Cr3+-E0индикатора.
ЕFe3+/Fe2+=E0Fe3+/Fe2++0.059/n(1)*lg*[Fe3+]/[Fe2+].
19.Теоретические основы трилонометрического метода анализа. Приготовление ст. р-ра в-ва комплексона(III). Установка точной конц-ии ст. р-ра в-ва трилона Б. применение трилонометрического метода анализа.
Метод основан на реакциях комплексообразования. К простейшим комплексонам относятся производные аминокарбоновых к-т: комплексон I, II и III – двунатриевая соль этилен диамин тетроуксусной к-ты. Na2 [H2Тр.Б]. метод на применение аминокарбоновых к-т и их солей. Одна молекула комплексона III реагирует только с одним ионом металла (не зависимо от степени ок-ия), т.к. 2 иона трилона Б замещается на металл любой степени ок-ия. Ме3++ Na2 [H2Тр. Б]→Na2[MeТр.Б]+2Н++2Na+, Ме2+ или Ме4+.Z=2.
Z комплексона равно 2, т.к. выделяется при реакции 2 иона водорода.
Точку экв-ти устанавливают с помощью индикаторов. Некоторые индикаторы не устойчивы при хранении, поэтому применяют свежеприготовленные р-ры или сухие индикаторные смеси. Наиболее часто используют индикаторы: кислотный хром тёмно-синий и эриохром чёрный или хромоген, мурексид.
Приготовление ст. р-ра в-ва комплексона III. Комплексон III (трилон Б) не отвечает требованиям, предъявляемым к исходному в-ву. Ст. р-р готовят из приблизительной навески. Рассчитать массу навески комплексона III, необходимую для приготовления р-ра с С(1/z)=0,05моль/л, V=0,2л. Массу навески взвесить на тех. весах, растворить в дист. воде. Объём воды отмерить измерительным стаканом.
m(Тр. Б)=V(р-ра)*С(1/z)*М(1/zТр. Б).
Установление точной конц-и ст. р-ра трилона Б. Устанавливают по ст. р-ру в-ва сульфата магния MgSO4 с молярной конц-ей 0,05моль/л. Взять аликвоту сульфата магния 0,01л, перенести в колбу для титрования, примерно вдвое разбавить водой, 0,005л аммиачной буферной смеси, 7-8 капель индикатора хромогена синего, титровать ст. р-ром трилона Б до перехода винно-красной окраски в синюю.
С(1/2 Тр.Б)=V(MgSO4)*С(1/2 MgSO4)/V(Тр. Б)=моль/л.
Применяется в заводских лабораториях, определения содержания многих Ме, применяется для анализа доломита СаСО3 и МgСО3, для смягчения воды – жёсткость воды обуславливается содержанием Са2+ и Мg2+. При добавлении трилона Б получается комплексное соединение и вода становится мягче. Для определения жёсткости воды. В теплоэнергетике для отмывки накипи в трубах, котлах. В медицине добавляют к крови, способствующей сохранности крови, диагностика раковых заболеваний ( в крови нет цинка), для смыва радиоактивных в-в.
ФХМА.
1.Сущность фотометрического анализа и область применения. Основной закон светопоглощения. Пропускание и оптическая плотность. Молярный коэф-нт светопоглощения, его физический смысл и значение.
Фотометрический метод анализа основан на избирательном поглощении (абсорбции) света анализируемым р-ром.в основе фотометрии лежит закон Бугера – Ламберта - Бера А=ЕСL, где А – оптическая плотность; Е – коэффициент поглощения, индивидуален для каждого соединения и показывает чувствительность метода; L – толщина слоя, кюветы; С – конц-ия в-ва. Закон: оптическая плотность р-ров при прочих равных условиях прямо пропорциональна конц-ии в-ва и толщине поглощающего слоя.
Молярный коэф-нт. Численное значение молярного коэф-та поглощения равно оптической плотности такого р-ра, конц-ия которого равна 1 грамм-моле (моль) в 1 л, при толщине поглощающего слоя в 1см. МКП не зависит от конц-ии в-ва при прохождении света данной длины волны. Величины МКП различны для р-ров разных соединений и колеблются в широких пределах: от единиц до сотен тысяч. МКП поэтому является мерой чувствительности колориметрических реакций. Чем больше величина МКП, тем выше чувствительность колориметрического определения. МКП можно рассчитать по результатам измерения оптической плотности р-ра данной конц-ии. Е=А/СL.
2.Спектры поглощения р-ров, их характеристика. Выбор спектральной области для фотометрического анализа. Светофильтры, их выбор.
Фотоколориметрия – анализ на основе измерения поглощения видимого света без предварительного выделения монохроматического излучения (здесь применяют белый свет непосредственно или пропущенный через широкополосные светофильтры).
Зависимость светопоглащения от длины волны излучения выражается кривой (спектром) поглощения (абсорбции) света данным в-вом. Спектр поглощения может быть представлен в виде графика, на котором по оси абсцисс откладывают длины волн ( в миллимикронах или микронах) или волновые числа (величины, обратные длинам волн, выраженные в обратных см – см-1). Ординатами спектра поглощения могут быть оптические плот-ти (А), логарифмы оптических пл-ей, молярные коэф-ты поглощения (Е) или логарифмы молярных коэф-ов пог-ия.
Спектр поглощения хар-ся наличием в нём определённого числа полос. Каждая полоса хар-ся в свою очередь положением максимума и выражается соответствующей длиной волны – λmax или волновым числом ωmax.
Спектр поглощения является индивидуальной хар-кой данного в-ва. На изучении спектров поглощения основан качественный анализ поглощающих свет в-в. Количественный анализ по светопоглощению основан главным образом на использовании закона Б-Л-Б: А=εСl.
Выбор. При колич-ом анализе по светопоглощению необходимо выбрать определённую длину волны поглощаемого излучения (соответствующий светофильтр, положение монохроматора). Выбранная длина волны должна отвечать нескольким требованиям: 1.высокая чувствительность рецептора (глаза, фотоэл-та) к выбранной длине волны. 2.воспроизводимость результатов при небольших отклонениях длины волны применяемого излучения (плоские максимумы на спектрах поглощения). 3.соблюдение закона Б-Л-Б. 4.во всех случаях измерение оптической плотности р-ра следует проводить при длине волны (λmax), которая соответствует максимальному поглощению света исследуемым р-ром. При этом достигается наибольшая чувс-ть и точность определения.
Обычно монохроматический пучок света с λmax выделяют с помощью монохроматоров, которые являются составной частью спектрофотометров. В случае фотоколориметров и фотометров приближённо монохроматический пучок света получают с помощью светофильтров, которые пропускают сравнительно узкую область спектра. При колориметрировании стремятся выбрать светофильтр, пропускающий свет в такой области спектра, в какой преимущественно поглощает свет соединение анализируемого эл-та, т.е. минимум поглощения светофильтра должен совпадать с максимумом поглощения р-ра. Когда спектр поглощения исследуемого р-ра неизвестен, то подбирают такой светофильтр, чтобы его окраска была дополнительной к окраске р-ра, например для жёлтых р-ров используют синий или фиолетовый светофильтр, для красных – зелёный и синий.
3.Количественный фотометрический анализ: метод градуировочного графика, метод сравнения оптических плотностей стандартного и исследуемого окрашенных р-ров, метод добавок, расчётный.
Метод градуировочного графика. Измеряют оптические плотности 5-10 р-ров с известной конц-ей. Строят калибровочный график, откладывая по оси ординат оптическую плотность, а по оси абсцисс – конц-ию. В случае подчинения светопоглощения р-ров закону Б-Л-Б все три точки укладываются на одну прямую. Затем измеряют оптическую плотность исследуемых р-ров и по градуировочной кривой находят их конц-ии. Этот метод наиболее удобен для серийных определений.
Метод сравнения оптических плотностей стандартного и исследуемого р-ров. Измеряют оптические плотности ст. и иссл-ого р-ров при одной и той же толщине поглощающего слоя (Lх=Lст). из уравнения Б-Л-Б следует Сх/Сст=Ах/Аст. по этому соотношению легко можно вычислить неизвестную конц-ию Сх.
Метод добавок. Измеряют оптическую плотность иссл-ого р-ра. Затем к нему прибавляют известное кол-во определяемого в-ва и снова измеряют оптическую плотность. Неизвестную конц-ию опред. в-ва находят путём сравнения оптической плотности исследуемого р-ра и его же с добавкой: Ах=ЕСхL, где Ах – оптическая плотность исследуемого р-ра. Ах+ст=ЕL(Сх+Сст), где Ах+ст – оптическая плотность исследуемого р-ра с добавкой. Отсюда следует: Сх=СстАх/Ах+ст-Ах.
4.Определение высоких конц-ий в-ва методом дифференциальной фотометрии.
При определение конц-ых р-ров наблюдается отклонение от линейной зависимости от закона Б-Л-Б. Для таких определений используется метод дифференциальной фотометрии. Сущность метода: оптические плотности исследуемого и стандартного окрашенных р-ров измеряются не по отношению к чистому растворителю с нулевым поглощением, а по отношению к окрашенному р-ру определяемого компонента с конц-ей С0 близкой к конц-ии исследуемого р-ра. Существует три варианта метода.
1.Конц-ия р-ра сравнения меньше конц-ии исследуемого р-ра.
Серия ст. р-ров С0, С1, С2, С3, Сn.
Исслед. р-р Сх, С0<Сх.
Измеренная экспериментально относительная оптическая плотность Аотн – это разность оптических плотностей фотометрируемого р-ра и р-ра сравнения.
Ахотн=Ах-А0=ЕL(Сх-С0), Астотн=Аст-А0=ЕL(Сст-С0)
Конц-ию исслед-ого р-ра определяют с помощью градуировочного графика или расчётным способом. Графический. Для построения градуировочного графика готовят серию ст. р-ров с конц-ей С1<С2<С3<Сn и измеряют их оптическую плотность по отношению к окрашенному р-ру сравнения с С0. Строят график. Принимая за начало отчёта конц-ию р-ра сравнения С0. Измерив Ахотн по графику находим Сх. При расчётном способе: Ахотн к соотношению /Астотн=(Сх-С0)/(Сст-С0) следовательно Сх=С0+Ахотн(Сст-С0)/Аст или Сх=С0+FАхотн. Метод удобен при измерении р-ра А>1.
2.Конц-ия р-ра сравнения больше, чем конц-ия исследуемого р-ра С0>Сх (обратное дифференцирование). Анализируемые р-ры условно принимают за р-ры сравнения и по отношению к ним измеряют оптическую плотность р-ра сравнения. Расчёт такой же только Ахотн=А0-Ах.
3.Двухстороннее или полное дифференцирование. Это сочетание прямого (С0<Сх) и обратного (С0>Сх) порядка измерений. При объединении этих двух методов значительно расширяются возможности определения. При фотометрировании исслед-ых и ст. р-ров, конц-ии которых больше, чем конц-ии р-ра сравнения, значения Аотн со знаком +, если конц-ия фотометрируемых р-ров меньше, чем конц-ия р-ра сравнения, то Аотнсо знаком -.
Для построения градуировочного графика готовят несколько р-ров с конц-ми меньше, чем р-ра сравнения и столько же ст. р-ров с конц-ми больше, чем р-ров сравнения. Для расчётного способа используются теже формулы в зависимости от того в какую область попала Ах.

5.Сущность потенциометрического анализ. Электроды первого, второго рода, окислительно-восстановительные. Примеры. Уравнение Нернста.
Потенциометрический метод анализа основан на зависимости потенциала от содержания в-ва. Эта зависимость выражается уравнением Нернста: Е=Е0+0,059/n*lgС,
RT/nF*ln=0.059, зависит от температуры.
Для ПА нужно составить электрохимическую ячейку из двух электродов: индикаторный и сравнения. Индикаторный – эл-д, потенциал которого зависит от конц-ии определённых ионов. Сравнительный – эл-д, потенциал которого не зависит от состава р-ра. Все эл-ды подразделяются на эл-ды первого рода, второго рода и о-в. Первого рода – металл опущенный в р-р его соли (Сu/CuSO4, Zn/ZnSO4). ЕI=Е0Ме0/Меn++0,059/n*lgСМе.
Второго рода – металл покрытый его трудно растворимой солью и опущенный в р-р с тем же анионом (хлорсеребряный, каломельный). Ag/AgCl, KCl – хлорсеребряный эл-д.
ЕII=E0-0.059/n*lgСCl-, зависит от аниона внутреннего р-ра.
Окислительно-восстановительные – пластинка инертного Ме, опущенная в р-р, где имеются окислительная и восстановительная формы (платиновый). Pt/Fe2+;Fe3+. Индикаторный эл-д выбирается либо по типу реакции либо от природы ан-ого в-ва.
Ео-в=Е0Fe2+/Fe3++0,059/1*lgС, окислительной формы/восстановительной формы.
6.Индикаторные электроды в потенциометрии. Классификация. Примеры. Уравнение Нернста. Выбор индикаторных электродов. Примеры. Электроды сравнения.
Потенциометрический метод анализа основан на зависимости потенциала от содержания в-ва. Эта зависимость выражается уравнением Нернста: Е=Е0+0,059/n*lgС,
RT/nF*ln=0.059, зависит от температуры.
Индикаторный – эл-д, потенциал которого зависит от конц-ии определённых ионов. Подразделяются на металлические и ионоселективные. Пример. Окислительно-восстановительные – пластинка инертного Ме, опущенная в р-р, где имеются окислительная и восстановительная формы (платиновый). Pt/Fe2+;Fe3+. Индикаторный эл-д выбирается либо по типу реакции либо от природы ан-мого в-ва.Ео-в=Е0Fe2+/Fe3++0,059/1*lgС, окислительной формы/восстановительной формы.
Ионоселективные (мембранные) эл-ды, в основе которых лежат ионообменные процессы, протекающие на границе мембран с р-ром электролита. Они подразделяются на эл-ды со стеклянной, твёрдой и с жидкой мембраной.
Стеклянный эл-д. (смотри вопрос 7).
Эл-д с твёрдой мембраной. Например, фторид селективный. Его мембрана состоит из моно кристалла лантан фтор 3 LaF3. 1- пластинка LaF3 (мембрана); 2- внутренний ст. р-р NaF+NaCl; 3- внутренний эл-д сравнения. Е=Е0-0,059/n*lgCF-.
Эл-д с жидкой мембраной. Например, нитратный эл-д. жидкая мембрана – это р-р электродно - активного в-ва в органическом растворителе, не смешивающимся с водой. Органическая и водная фазы отделены полупроницаемой инертной мембраной. Недостаток: короткое время жизни эл-да.
7.Стеклянный ионоселективный эл-д. Устройство. Уравнение Нернста. Особенности. Измерение рН р-ров со стеклянным и хлорсеребряным эл-дом.
Стеклянный эл-д – индикаторный эл-д, его потенциал зависит от конц-ии ионов Н+ в р-ре. Е=Е0+0,059/n*lgCН+, рН=-lgСН+, -рН=lgСН+; n=1, Е=Е0-0,059рН.
Поэтому используется для определения кислотности (рН). Мембрана (шарик) такого эл-да изготовлена из специального стекла, содержащего Na2O, СаО, SiO2.
Если стек-ый эл-д длительное время выдержать в воде, то на обеих поверхностях мембраны образуется тонкий слой 10-4мм гидротированного геля. Все пустоты занимаются ионами Н+, вытеснившими находившиеся там ионы Na+, таким образом потенциал эл-да зависит от конц-ии ионов Н+. 1- стеклянная мембрана, 2- внутренний р-р HCl, 3- серебряная проволока. В последнее время разработано большое кол-во различных стеклянных мембран, в состав которых введены другие в-ва (Ва, Li и т.д.) поэтому с помощью таких стеклянных эл-дов можно измерять не только ионы Н+, но и Na+, К+, NH4+, Li+, Ag+, РЗМ.
В паре с хлорсеребряным эл-дом стеклянный эл-д используют при измерении рН р-ра (титруем). Используется рН метр.
Потенциометрический метод анализа основан на зависимости потенциала от содержания в-ва. Эта зависимость выражается уравнением Нернста: Е=Е0+0,059/n*lgС,
RT/nF*ln=0.059, зависит от температуры.
8.Потенциометрическое титрование. Сущность метода. Кривые титрования. Способы определения точки экв-ти. Потенциометрическое титрование в методе нейтрализации: измерение рН и потенциала стеклянного эл-да, кривые титрования. Потенциометрическое титрование в методе о-в, изменение потенциала индикаторного эл-да. Кривые титрования.
Потенциометрический метод анализа основан на зависимости потенциала от содержания в-ва. Эта зависимость выражается уравнением Нернста: Е=Е0+0,059/n*lgС,
RT/nF*ln=0.059, зависит от температуры.
Потенциометрический метод анализа делится на прямую (ионометрию) потенциометрию и косвенную – потенциометрическое титрование. Разновидность титриметрического метода (бюретка, титрант), в котором точка экв-ти устанавливается не визуально (не по индикатору), а по прибору. Анализируемый р-р титруем и при этом замеряем по прибору значение потенциала. График зависимости от Vтитранта Е=f(Vтитр).
Иногда по интегральной кривой точку экв-ти определить сложно, тогда строят дифференциальную прямую.
В методе нейтрализации строится график зависимости рН=f(Vтитр), кривые выглядят также.
Для титрования используем индикаторный эл-д (стеклянный), а хлорсеребряный как сравнение.
О-в строим Е=f(Vтитр), график такой же, эл-д платиновый, сравнение – хлорсеребряный.
9.Сущность электролиза. Химические поцессы при электролизе. Объединённый закон электролиза. Выход по току.
При электролизе р-ра в-во разлагается под действием эл. тока на эл-дах происходит выделение составных частей электролита или водорода и кислорода из воды. Выделение Ме на катоде зависит от их стандартных потенциалов (Е0):
1.если в-во в ряду напряжений находится до алюминия включительно:
К=2Н2О+2е→Н2++2ОН-
2.если Ме стоит в интервале отAl до Н то: К= Zn2++2e→Zn0, 2Н2О→Н2+2ОН-.
3.если в-во стоит после Н, то: К=Cu2++2e→Cu0.
На аноде. 1.Если анион является остатком без кислородной кислоты, то он и будет реагировать: А 2Cl—2e→Cl2. 2.Если анион является остатком кислорода, содержащей кислоты SO42-, NO3, РО4, тогда: А 2Н2О-4е→4Н2++2О2. 3.Если гидроксид ион:
А 4ОН-4е→2Н2О+О2.
Согласно законам Фарадея масса электрохимически окисленного или восст-ого в-ва равна: m=M*Q/nF, где Q – кол-во электричества равное I*t. m=MIt/nF.
10.Кулонометрическое титрование, сущность метода. Ячейка для КТ, устройство. Реакция электрогенерирования титранта. Визуальные и инструментальные способы индикации точки экв-ти. Определение времени титрования. Вычисление результатов анализа.
Кулонометрия основана на измерении кол-ва электричества, затраченного на электропревращение определяемого в-ва (прямая кулонометрия) или на получение титранта, реагирующего с определяемым в-вом (косвенная кулонометрия).
m=M*Q/nF, где Q – кол-во электричества равное I*t, n – число эл-нов, участвующих в превращении; F – постоянная Фарадея, 965000Кл/моль.
Метод прямой кулонометрии применяют для определения только электроактивного в-ва, поскольку он основан на непосредственном электропревращении этого в-ва. Измерение можно проводить либо при постоянной силе тока, либо при постоянном потенциале рабочего эл-да. Косвенная кулонометрия (кулонометрическое титрование) применяется чаще, так как этот вариант пригоден для определения и электроактивных, и электронеактивных в-в. Титрант для кулонометрического титрования получают на рабочем или генераторном эл-де из вспомогательного реагента (например, в результате окисления I- до I2), из растворителя (например, в результате восст-ия воды до ОН- ионов), материал электрода (например, в результате ок-ия Ag до Ag+). По мере образования титрант вступает в реакцию с анализируемым в-вом, например, титрант I2 взаимодействует с тиосульфатом натрия по реакции: 2Na2S2O3+I2→Na2S4O6+2NaI.
Для установления конечной точки титрования используются визуальные и инструментальные методы (потенциометрия, амперометрия). Сила тока должна быть постоянной в течение электролиза. Массу определяемого в-ва рассчитывают по формуле: m=M*Q/nF, где Q – кол-во электричества равное I*t, то Кл=А*с; m=MIt/nF, где I – сила тока, А; t – время электролиза, с.
Ячейка для КТ содержит рабочий эл-д и вспомогательный, отделённый от рабочего полупроницаемой мембраной (пористое стекло, целлофановая плёнка). Обычно его помещают в сосуд с пористым дном, внутрь которого заливается подходящий электролит (KCl,KSO4). Это в случае применения визуального способа индикации т.э. Если используют инструментальные способы индикации, например, потенциометрический, то, кроме рабочего эл-да, в анализируемый р-р опускают ещё два эл-да – индикаторный и сравнения.
Основное преимущество КТ – не нужно готовить титрант заранее, стандартизировать его и хранить. С помощью одного и тогоже источника тока можно получать любые титранты, в том числе и неустойчивые.
Выход по току - это отношение кол-ва электропревращённого в-ва к теоретически вычисленного по закону Фарадея: =m(практич)*100%/m(теоретич).
11.Сущность вольтамперометрического анализа. Полярография с ртутным капающим эл-дом. Схема полярографа. Получение полярограммы, её объяснение.
Вольтамперометрический метод анализа основан на регистрации и изучении зависимости силы тока, протекающего через электролитическую ячейку, от внешнего наложенного напряжения. Графическое изображение этой зависимости называют вольтамперограммой. Анализ вольт-мы даёт информацию о качественном и количественном составе анализируемого р-ра. Для регистрации вольт-мм используют электролитическую ячейку, состоящую из индикаторного эл-да и эл-да сравнения – насыщенный каломельный эл-д или слой ртути на дне электролизера (донная ртуть).
Электрохимический метод анализа, в основе которого лежит зависимость между хар-ром поляризации рабочего эл-да и составом р-ра, в котором он находится, называется полярографией. Само слово полярография означает запись процесса поляризации.
В электролизер, содержащий анализируемый р-р помещается Ме ртуть, которая явл-ся анодом. Катодом служит ртутный капающий эл-д. Капилляр этого эл-да погружён в анализ-ый р-р. Через электролизер протекает постоянный ток, напряжение которого можно изменять с помощью реохорда и измерять гальванометром его силу. В ртуть вводят контактный провод, подключённый к источнику постоянного тока, поэтому капля ртути на кончике капилляра до момента её отрыва явл-ся эл-ном (чаще всего катодом). Скорость капания ртути должна быть равномерной и составлять одну каплю за 3-5 сек.
Поверхность ртути на дне электролизера больше поверхности капли катода в несколько тысяч раз. При прохождении небольших по величине токов потенциал данной ртути остаётся постоянным и эл-д не поляризуется. Приложенный к ячейке напряжение рассчитываем по формуле: Е=φа-φк+IR, где φа – потенциал, анода, φк – потенциал катода, R – сопротивление р-ра. Несмотря на высокое напряжение потенциал анода во время эл-за остаётся постоянным, т.к. на его большой поверхности создаётся малая сила тока и поэтому изменение конц-ии эл-та при анодном слое не значительно. Е=-φк, φа=const, IR=мала. В качестве неполяризующегося эл-да можно применять каломельный эл-д с большой поверхностью.
12.Помехи, искажающие полярографическую волну. Мешающее влияние растворённого кислорода, его устранение в нейтральных, щелочных и кислых средах. Полярографические максимумы первого и второго рода, их устранение.
Кислород восст-ся на ртутном катоде даёт две волны, т.к. восст-ся в две стадии: О2+2Н++2е→Н2О2 1 стадия, Н2О2+2Н++2е→2Н2О 2 стадия. Так как кислород восст-ся раньше других катионов, то происходит искажение волны, что мешает определению исслед-ого в-ва. Особенно сильно кислород мешает опред-ию Ме, потенциал которых близок к 0 (медь, сурьма, свинец, кадмий). Кислород следует удалять из р-ра. Если иссле-ый р-р имеет щелочную или нейтральную среду, то кислород устраняется легко, к р-ру прибавляют Na2SO3+1/2О2→Na2SO4. Если исслед-ый р-р имеет кислую среду, то процесс идёт сложнее. Перед полярографированием в течение 20 минут пропускают газ (Н2, СО2, N2). Газы удаляют кислород из р-ров.
Максимумы 1 и 2 рода. В области предельного диффузионного тока могут возникать мак-мы различной формы, которые искажают волну и затрудняют измерение высоты полярограммы. Максимумы разделяются на первого и второго рода.
1 рода. Вызван неравномерной поляризацией ртутной капли. Такие мак-мы наблюдаются для сильно разбавленных р-ров. В нижней части ртутной капли скапливается больше зарядов, чем в верхней. При этом капля стремится выровнять свою поверхность натяжения на всех участках и начинается давление ртути снизу вверх (катод), сверху вниз (анод). В результате таких движений происходит перемешивание ближайшего к капле слоя р-ра, ток увеличивается. Мак-м 1 рода устраняют с помощью поверхностно активными в-ми, которые тормозят движение поверхности ртути и явл-ся диффузионными. В качестве ПАВ использовать желатин, столярный клей (ПАВ – поверхностно активные в-ва).
2 рода. Появляется при работе с быстро капающими капиллярами при высоких конц-ях эл-тов (выше, чем 0,1 моль). Струя ртути о ндо разрывается так, что появляются вехревые струйки ртути, которые приводят в движение всю поверхность капли. И увлекают за собой прилегающие слои р-ра. Происходит перемешивание, повышается ток. Максимум 2 рода имеет более сильную форму, чем мак-м 1 рода. Уменьшают с помощью замены капилляров, с уменьшением скорости капания и применяется ПАВ.
13.Миграционный ток. Мешающее влияние миграционного тока в полярографии. Р-р фон, его состав и назначение. Примеры. Потенциал полуволны, факторы, влияющие на его величину. Полярограмма смеси ионов. Качественные определения в полярографии.
Восст-щиеся или ок-щиеся ионы в отсутствии постороннего эл-та достигают поверхности эл-да под действием 2-х факторов: диффузии и миграции. Миграция – перемешивание ионов под действием электростатического поля катода. В следствии миграции кол-во катионов, (10-2%) поступающих к катоду в ед-цу времени, увеличивается и предельный ток возрастает. Iпред=Iдиф+Iмигр. Миграционный ток может значительно исказить вид полярограммы. Ионы фона располагаются у поверхности эл-да. Электрическое поле эл-да этими ионами не распространяется в глубину р-ра. Кол-во восст-ся ионов, перемешивающихся под действием поля, ничтожно мало по сравнению с кол-вом диффундирующих ионов.
В качестве фона применяют соли щелочных, щелочноземельных Ме, соли аммония, гидроксид аммония, щёлочи, кислоты при конц-ии в 100-1000 раз превышающей конц-ию определяемого в-ва. Фон значительно увеличивает электрическую проводимость анализируемого р-ра.
В основе качественного поляр-ого анализа лежит измерение потенциала полуволны. U1/2 – потенциал соответствующий середине поляр-ой волны.
зависит от природы в-ва ,от состава среды (фона). φв=φ0+0,059*lgС/n.
Иссслед-ый р-р поляр-ют и регистрируют силу тока, по данным чертят поляр-му, на графике находят потенциал полуволн, найденные полуволны учитывают фон и сравнивают с табличным и определяют к каким в-вам они соответствуют.
Табличные данные и приводятся в справочниках по аналитической химии и литерату-
ре поляр-ии. Данные делются относительно каломельного эл-да. При анализе учитывают, что элементы дают раздельные волны, если разница в потенциалах полуволн составляет не менее 0,2В, иначе они сольются. Некоторые ионы (2,3 валентные) могут дать несколько волн: Cu в аммиачном р-ре (2 волны) нижняя - Cu2++1е=Cu+, - верхняя Cu++1е=Cu0. количественное определение в данном примере находится по верхней волне.
14.Уравнение Ильковича для предельного диффузионного тока. Количественный полярографический анализ. Метод градуировочного графика, метод сравнения, метод добавок.
Для количественного определения в-ва используется прямо пропорциональная зависимость между силой предельного диффузионного тока и конц-ей в-ва. Эта зависимость выражается различными уравнениями для разных типов используемых эл-дов и для случая ртутно-капельного эл-да носит название уравнения Ильковича:
Iпр=605nД1/2m2/3t1/6C, где iпр – сила предельного диффузионного тока, мкА; n – число эл-нов, участвующих в электрохимической реакции, С – конц-ия определяемого в-ва, ммоль/л; D – коэффициент диффузии ионов, см2*сек-1; m – масса ртути, вытекающей из капилляра в 1 сек, мг; t – время образования одной капли или время жизни, сек.
При полярографировании создают условия, при которых величины m и t остаются постоянными. Тогда все постоянные величины можно объединить в одну постоянную К и получить следующее уравнение: iпр=КС. Заменим величину силы тока Iпр на пропорциональную ей величину h. Получим уравнение: h=КС, где h – высота волны; К – коэф-нт пропорциональности; С – концентрация.
Метод градуировочного графика. Полярографируют ряд ст. р-ров определяемого эл-та, измеряют их высоты волн. По полученным данным строят градуировочный график в координатах высота волны – содержание или конц-ия компонента. Полярографируют анализируемый р-р в тех же условиях, измеряют высоту волны и по графику находят неизвестное содержание определяемого в-ва.
![]()
![]()
Метод стандартов. В совершенно одинаковых условиях снимают полярограмму анализируемого р-ра, а затем полярограммы 2-3 ст. р-ров, подобранных в такой конц-ии, чтобы полученные высоты волн при той же чув-ти гальванометра были примерно равны высоте волны, полученной при полярографии анализируемого р-ра.
Для анализируемого р-ра: hх=КСх.
Для ст-ого р-ра: hст=КСст. разделим одно уравнение на другое hх/hст=Сх/Сст; Сх=Сстhх/hст.
Метод добавок. Метод может быть выполен расчётным или графическим способом. Расчётный способ состоит в следующем: измеряют высоту волны анализируемого р-ра. Далее измеряют высоту волны этого же р-ра с добавкой некоторого известного кол-ва в-ва. Значения высот волн будут равны: hх=КСх, hх+доб=К(Сх+Сдоб), где hх+доб – высота волны анализируемого р-ра с добавкой; Сдоб – конц-ия добавки в анализируемом р-ре. Составим пропорцию: (hх+доб-hх)-Сдоб и hх-Сх, тогда Сх=Сдоб*hх/hх+доб-hх.
Графический способ состоит в следующем: измеряют высоту волны анализируемого р-ра hх. затем измеряют высоты волн анализируемого р-ра с добавками: hх+доб1, hх+доб2, hх+доб3. строят градуировочный график в координатах высота волны – конц-ия добавок. Проводят по точкам прямую до пересечения с осью абсцисс левее нуля. Полученный отрезок даёт Сх.
15.Распределительная жидкостная колоночная хроматография. Техника выполнения. Твёрдые носители, их роль, примеры. Жидкие неподвижные фазы, требования к ним, примеры. Подвижный растворитель, требования к нему, назначение. Примеры. Механизм разделения, коэффициент распределения, его значение.
Распределительная хр-ия основана на различии коэффициентов распределения отдельных компонентов между двумя несмешивающимися растворителями. В методе используется твёрдый носитель, который распределяет жидкую фазу на большой поверхности (т.е. его пропитывают неподвижным растворителем). В качестве твёрдого носителя используют пористые в-ва, например, силикагель, крахмал, целлюлоза. В качестве неподвижного растворителя используют полярные жидкости (вода, серная кислота, метиловый спирт).
Требования к неподвижным фазам. 1. полная химическая инертность по отношению к компонентам разделяющим смесям и твёрдому носителю. 2. высокая селективность. 3.малая вязкость. 4. малая летучесть. 5. термическая устойчивость. 6. прочное удерживание на твёрдом носителе.
Подвижный растворитель, требования к нему. В качестве подвижного растворителя используют хлороформ, это должны быть менее полярные жидкости несмешивающиеся с неподвижным растворителем.
Сначала оба раст-ля насыщают друг другом. Разделяемую смесь в-в растворённую в подвижном раст-ле вводят в колонку и начинают промывать через колонку чистым подвижным раст-лем. При промывании в-ва смеси непрерывно перераспределяются между двумя несмешивающимися фазами. Т.к. коэффициенты распределения различных компонентов смесей различны, скорость передвижения отдельных компонентов неодинакова. К=Сподвижн/Снеподвижн, где Спод – это конц-ия растворённого в-ва в подвижной фазе; Снепод – это конц-ия раст-ого в-ва в неподвижной фазе.
Носитель СаSO4, растворитель – сложный эфир, зелёный – хлорофилл, оранжевый – α каротин, жёлтый – β каротин.
16.Ионообменная хроматография. Сущность ионного обмена. Ионообменники, их классификация, примеры. Реакции ионного обмена. Основные св-ва ионитов. Подготовка ионитов. Применение ионного обмена в аналитической химии, примеры.
Ионный обмен – это химическое взаимодействие активных групп твёрдой фазы с ионами в р-ре. В качестве такой фазы используются ионообменники (сорбенты – твёрдое нерастворимое в-во, иониты – вступают в обмен с катионами – катионитами, а с анионами – анионитами).
Ионообменники: 1.природные (глина, угли, силикагель); 2.синтетические (ионообменные смолы): а) катиониты КУ-1 (марка) бывают сильно кислотные и слабо кислотные; б) аниониты An 21 , бывают сильно и слабо основные.
RSO3H+NaCl↔RSO3Na+HCl (с катионом).
После реакции катионит регенерируют (восст-ют), т.е. обрабатывают кислотой, и реакция идёт в другом направлении. RNH3OH+NaCl↔RNH3Cl+NaOH (с анионами).
Перед началом работы сухой ионит замачивают для набухания в воде, далее переводят при необходимости в кислотную или щелочную форму. Ионный обмен применяется для разделения смеси, для очистки, для конц-ии определяемого компонента.
17.Количественный анализ в жидкостной колоночной хроматографии. Хроматограмма. Способы определения высоты, ширины и площади пика. Метод нормировки, метод нормировки с поправочным коэффициентом, метод градуировочного графика, метод внутреннего стандарта.
Для количественного определения отдельных в-в или ионов хроматографическую колонку промывают подходящим растворителем (например, р-ром кислоты), постепенно вымывая из неё одну за другой все образовавшиеся зоны и, собрав отдельные порции, стекающие из колонки жидкости определяют в них соответствующие в-ва или ионы. В анализе используют также осадочную хроматографию, при которой разделение ионов на колонке основано на различной растворимости соединений. Большое значение для анализа неорганических соединений имеет ионообменная хр-ия.
Метод нормировки. МН основан на том, что сумму площадей всех пиков на хроматограмме принимают за 100%. Массовую долю анализируемого в-ва рассчитывают по формуле: ω%=S1*100%/S1+S2+S3, где S1 – площадь искомого пика; S1+S2+S3 – сумма площадей всех пиков; ω% - массовая доля в-ва. МН с поправочным коэф-ом. Для учёта различия в чувствительности детектора для каждого компонента смеси определяют экспериментально поправочные коэф-ты. Тогда расчёт с учётом поправочных коэф-тов будет таким: ω%=K1S1*100%/K1S1+K2S2+…+KnSn, где К1, К2…Кn – поправочные коэф-ты.
Метод градуировочного графика или абсолютной калибровки. В методе строят градуровочный график зависимости площади или высоты пика от конц-ии в-ва. Измеряют площадь пика анализируемого в-ва в смеси и по графику находят его конц-ию. Метод является основным в определении микропримесей.
Метод внутреннего стандарта. Метод основан на введении в анализируемую смесь точно известного кол-ва стандартного в-ва. После хроматографирования измеряют площадь пиков анализируемого и ст. в-ва. Массовую долю компонента рассчитывают по формуле: ω%=mвн.ст.*S1*100%/Sвн.ст.*m(пробы), где S1 – площадь пика анализируемого в-ва; Sвн.ст. – площадь пика внутреннего ст-та; m(пробы) – масса анализируемой пробы, г; mвн.ст. – масса внутреннего ст-та, г. С учётом поправочного коэф-та.
ω%= mвн.ст.*S1*К1*100%/ Sвн.ст.*m(пробы), где К1 – поправочный коэф-нт определяемого в-ва по отношению к внутреннему ст-ту.
18.Газоадсорбционная хр-ия. Техника выполнения, адсорбенты, газы-носители, примеры. Механизм разделения. Газожидкостная хр-ия. Техника выполнения, твёрдые носители, жидкие неподвижные фазы, требования к жидким неподвижным фазам. Газы-носители, их роль и назначение, примеры. Механизм разделения.
ГХ – метод разделения летучих соед-ий, основанный на распределении в-ва между двумя фазами (неподвижная и газ).
Газовая хроматография (ГХ):1.газоабсорбционная (ГАХ) 2.газожидкостная (ГЖХ)
Неподвижная фаза твёрдая(угли, силикагель); жидкая (орг.жидкости)
Подвижная фаза газ газ.
Объекты анализа в ГХ – газы, жидкости, переведённые в пар (термоустойчивые соединения).
ГАХ: распределение в-в в ГАХ между подвижной и неподвижной фазами опред. процессом абсорбции. Требования к абсорбентам. 1.большая удельная поверхность. 2.селективность к опред. компонентам. 3.химическая инертность. 4.однородность. 5.механическая прочность. В качестве подвижной фазы используется газ-носитель. Требования к газу-носителю. 1.должен обеспечивать полное разделение компонентов смеси. 2.должен соответствовать чувствительности и типу детектора. 3.инертным к разделяемым в-вам и материалу колонки. 4.быть химически чистым.
Выбор газоносителя обусловлен эффективностью хроматографической колонки, чувствительностью и принципам действия детектора. Пример, гелий, аргон, углекислый газ, воздух, инертный газ.
ГЖХ. Метод основан на распределении компонентов смеси между газоносителем и неподвижной жидкой фазой в зависимости от их избирательной абсорбности тонкой плёнкой жидкости, закреплённой на инертном твёрдом носителе (стеклянные шарики).
В качестве газа-носителя используются теже в-ва, что и в ГАХ. В качестве неподвижной жидкой фазы используют: неполярные (насыщенные углеводороды), умереннополярные (сложные эфиры), полярные (полигликоли).
Требования к ним. Селективность разделения за счёт различной растворимости компонентов, небольшая вязкость, химическая инертность, возможность образовать равномерную плёнку на носителе.
В качестве твёрдых носителей используются различные сорбенты, силикагель, стенки колонки.
19.Количественный анализ в ГХ. Способ определения площади пика. Метод нормировки, метод нормировки с поправочным коэф-м, метод абсолютной калибровки. Метод внутреннего стандарта.
В основе количественного анализа газовой смеси лежит определение основных параметров хроматографического пика: высота, ширина, площадь, время удерживания объёма.
Метод нормировки. МН основан на том, что сумму площадей всех пиков на хроматограмме принимают за 100%. Массовую долю анализируемого в-ва рассчитывают по формуле: ω%=S1*100%/S1+S2+S3, где S1 – площадь искомого пика; S1+S2+S3 – сумма площадей всех пиков; ω% - массовая доля в-ва. МН с поправочным коэф-ом. Для учёта различия в чувствительности детектора для каждого компонента смеси определяют экспериментально поправочные коэф-ты. Тогда расчёт с учётом поправочных коэф-тов будет таким: ω%=K1S1*100%/K1S1+K2S2+…+KnSn, где К1, К2…Кn – поправочные коэф-ты.
Метод абсолютной калибровки основан на использовании зависимости высоты или площади пика от кол-ва g соответствующего в-ва в смеси. Для этого строят калибровочный график в координатах h(Q)-g по хроматограммам известных в-в, взятых в различных, но точно измеренных кол-вах. Метод абсолютной калибровки достаточно прост, но точность его в значительной степени зависит от постоянства режима и тщательности приготовления и анализа стандартных смесей. Этот метод особенно широко применяется при определении одного или нескольких компонентов смеси. Является основным при определении микропримесей.
Метод внутреннего стандарта. Основан на введении в анализируемую смесь определённого кол-ва ст. в-ва. ω%=К1S1R*100R/KстSст, где К1 и Кст – поправочные коэф-ты к площадям пиков компонента и внутреннего ст-та, зависящие от чувствительности детектора; S1 и Sст – площади соответствующих пиков; R – отношение массы внутреннего ст-та к массе анализируемой смеси. Основная трудность метода заключается в выборе и точной дозировке ст. в-ва. В-во, используемое в качестве вн. ст-та, не должно входить в состав исследуемой смеси. Кроме того, его пик должен практически полностью отделяться от остальных пиков.
20.Принципиальная схема газового хроматографа. Основные узлы прибора, их назначения. Газы-носители. Система подготовки газа-носителя. Дозирующее устройство. Способы введения газообразных, жидких, твёрдых проб в хроматограф. Хроматографические колонки, их значение газоадсорбционной и газожидкостной хроматографии. Адсорбент. Твёрдые носители, жидкие неподвижные фазы. Механизм разделения на колонках. Детекторы, их устройство и принцип действия.
1.балон с газовым носителем. 2.испоритель. 3.система ввода пробы. 4.колонка (хроматографическая). 5.термостат. 6.детектор. 7система регистрации сигнала.
Термостат используется для поддержания постоянной температуры, т.к. сорбция зависит от температуры. Испаритель используется для перевода пробы в газообразное состояние. Хроматографические колонки, изготовленные из нержав. стали, бронзы, кварца (должен быть инертен). Колонки могут быть: спиралевидные, прямые, у-образные U, различного диаметра (в мм), различной длины (до нескольких метров). Детекторы. Чаще всего используются в качестве детекторов: каторометр (основан на сравнении теплопроводности газоносителя и смеси газоносителя с анализируемым компонентом); пламенноионизационный детектор (основан на ионизации органических горючих в-в в воздушно водородном пламени и измерении ионного тока); детектор электронного захвата (основан на ионизации газоносителя электронами). В качестве системы регистрации сигнала может быть самописец, компьютер на котором мы получаем хроматограмму (это графическая зависимость сигнала детектора от времени).
Похожие работы
... (II), (II)-(III) и(III). Гидроксиды и соли железа(II) и(III). Ферраты(III) и(VI). Комплексные соединения железа. Соли и комплексные соединения кобальта(II) и никеля(II). Органическая химия Характеристика каждого класса органических соединений включает: особенности электронного и пространственного строения соединений данного класса, закономерности изменения физических и химических свойств в ...
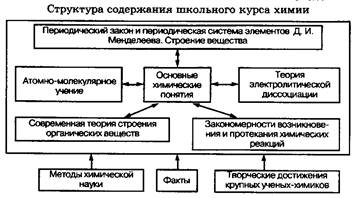
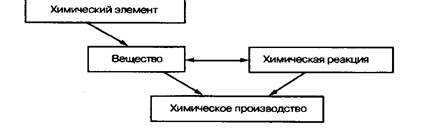
... ведущими являются понятия «вещество» и «химическая реакция». Именно этим понятиям принадлежит решающая роль при построении содержания различных курсов химии. Схема 2. Взаимосвязь систем важнейших химических понятий в курсе химии средней школы Отбор необходимых фактов для изучения химии превращается в сложную проблему. Постоянное стремление уменьшить описательный материал в курсе химии в ...
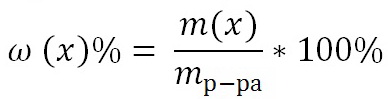
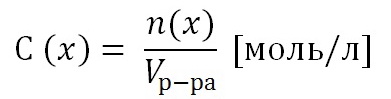
... с кислородом, восстановлением - отнятие кислорода. С введением в химию электронных представлений понятие окислительно-восстановительных реакций было распространено на реакции, в которых кислород не участвует. В неорганической химии окислительно-восстановительные реакции (ОВР) формально могут рассматриваться как перемещение электронов от атома одного реагента (восстановителя) к атому другого ( ...
... свежесорванных растениях массовая доля сухого вещества составляет (100–82,5)=17,5%. Тогда 100 г сырья – 17,5 г сух. раст. матер. х г сырья – 17,7 г сух. раст. матер. Отсюда х = 100•17,7/17,5 = 100,1 г. химия медицина курс Поскольку в настоях и отварах трав абсолютно точно дозировать содержание лекарственного вещества невозможно, с достаточной степенью точности можно принять, что необходимое ...
0 комментариев