Навигация
1.3.2. Физические методы
Основаны на решении пространственных структур РНК-белковых комплексов с атомарным разрешением. К этим методам относят метод ядерного магнитного резонанса (ЯМР) и метод рентгеноструктурного анализа.
Метод ЯМР позволяет видеть структуру макромолекулы в растворе без ограничений на взаимное расположение атомов, накладываемых кристаллической упаковкой. В настоящее время из-за сложности и величины РНК-белковых комплексов сфера его применения сильно ограничена.
Наиболее эффективным и универсальным методом определения трёхмерной структуры РНК-белковых комплексов с атомным разрешением является рентгеноструктурный анализ. Метод основан на свойстве биологических макромолекул образовывать монокристаллы, представляющих собой множество идентичных молекул, упорядоченных определенным образом. С кристаллов возможно получение рентгеновской дифракционной картины, с последующим анализом полученных данных и построении на их основе карты электронной плотности. Затем получают пространственную модель структуры, которая после ряда уточнений минимизируется по химической и кинетической энергиям [5].
Наряду с методом ядерного магнитного резонанса, рентгеноструктурный анализ также не лишён своих недостатков, наиболее значимыми из которых являются проблема кристаллизации РНК-белковых комплексов (необходимо наличие достаточно крупных, стабильных и однородных кристаллов), а так же фазовая проблема, решаемая методами изоморфного замещения, молекулярного замещения и аномальной дисперсии на нескольких длинах волн.
В последние годы, благодаря использованию синхротронного излучения, новых типов детекторов, а также разработке новых программ для обработки данных и расчёта фаз, метод рентгеноструктурного анализа получил наиболее широкое распространение.
2. Экспериментальная часть
2.1. Материалы и методы исследования
2.1.1. Метод молекулярного замещения
Для решения проблемы фаз в данной работе использовался метод молекулярного замещения. Метод основан на использовании известной структуры гомологичной молекулы (белка, РНК или ДНК) в качестве модели для получения начального приближения набора фаз [16].
Для расчета начального набора фаз необходимо, чтобы модель наилучшим образом аппроксимировала положения неизвестной молекулы в элементарной ячейке кристалла. Определение таких положений модели является основной задачей метода молекулярного замещения, которая обычно решается в два этапа. На первом этапе определяется ортогональное преобразование W, обеспечивающее правильную ориентацию модели в кристаллической ячейке. На втором этапе проводится поиск вектора трансляции v, задающего положение ориентированной модели в элементарной ячейке кристалла. Чаще всего, вышеупомянутое ортогональное преобразование выражается через углы Эйлера или сферические углы [37].
Функции вращенияПреобразование можно найти путем сравнения функций Паттерсона кристалла неизвестной молекулы Px и модели Pm. Критерием соответствия ориентации модели и неизвестной молекулы служит так называемая функция вращения, которая представляет собой интеграл перекрывания функций Px(r) и Pm(r) в элементе объема U и имеет максимумы если системы внутренних векторов модели и неизвестной молекулы ориентированы одинаково. Существуют методы расчета функции вращения как в прямом [22] так и в обратном пространстве [37]. В случае прямого пространства функции Паттерсона Px и Pm рассчитываются в явном виде при заданном разрешении с помощью преобразования Фурье. Затем происходит вращение Pm относительно Px с заданным шагом и ищутся максимумы функции вращения:
![]() , (1)
, (1)
где область интегрирования U - как правило сферический слой с центром в начале координат, задающийся минимальным и максимальным радиусами rmin и rmax, соответственно. Радиус rmin выбирается таким образом, чтобы исключить пик функции Паттерсона в начале координат (обычно rmin ³ 2Å), который может порождать ошибки при численном интегрировании. Радиус rmax выбирается так, чтобы включить в область интегрирования максимальное количество внутримолекулярных векторов при минимально-возможном количестве межмолекулярных [5].
Для уменьшения расчетных затрат при численном интегрировании на сетке используются только те точки, в которых функция Pm принимает наибольшие значения, а значения функции Px в этих точках рассчитываются с помощью процедуры интерполяции [22].
Применяя преобразование Фурье и теорему Парсеваля для уравнения (1) можно получить выражение для функции вращения в обратном пространстве [37]:
![]() , (2)
, (2)
где h = (h,k,l) обозначает миллеровские индексы, а - транспонированную матрицу оператора . Действие на |Fm(h)|2 будет в общем случае приводить к возникновению точек в обратном пространстве, которые не описываются целочисленными индексами (h,k,l). Значения |Fm(h)|2 в таких точках могут быть получены с помощью так называемой интерференционной функции G [2]:
![]() (3)
(3)
Анализ максимумов R() позволяет не только выявить наиболее вероятные ориентации модели в ячейке кристалла неизвестной молекулы, но и, в случае нескольких молекул в независимой части элементарной ячейки, найти операции точечной некристаллографической симметрии, связывающие ориентации этих молекул.
2.1.1.2. Функции трансляцииВторым этапом решения задачи молекулярного замещения является определение положения ориентированной молекулы в ячейке кристалла. Критерием соответствия положения модели и неизвестной молекулы служит функция трансляции. Существует много вариантов определения функции трансляции, в которых используются как функции Паттерсона [3, 36] так и коэффициенты корреляции между экспериментальными и расчетными амплитудами структурного фактора [20]. Функция трансляции может также включать фазовую информацию [4] и ограничения на возможную кристаллическую упаковку молекул [22]. Основной целью при этом является нахождение глобальных максимумов функции трансляции в зависимости от вектора трансляции v, описывающего положение модели в элементарной ячейке. Эта задача обычно решается с помощью процедуры поиска на сетке разбитой по компонентам вектора v.
Функция трансляции, в которой используется перекрывание между экспериментальной и расчитанной по модели функциями Паттерсона имеет общий вид:
 , (4)
, (4)
где Sj обозначает операторы симметрии данной группы [15].
Наличие экспериментальной фазовой информации может существенно повысить отношение сигнал-шум в пиках функции трансляции, даже если экспериментальный набор фаз содержит значительные ошибки (например, в тех случаях, когда имеется только одна изоморфная производная). В формулировке Рида и Ширбека [35] функция трансляции, включающая фазовую информацию, определяется следующим образом:
, (5)
где ρx и ρm – функции экспериментальной и модельной электронной плотности, соответственно.
Кроме правильной ориентации модели, к основным факторам влияющим на точность решения функции трансляции относятся качество и полнота модели и рентгеноструктурных данных, диапазон разрешений, а также критерий отбора в соответствии с которым те или иные структурные амплитуды включаются в расчет. Также как и для функции вращения, исключение слабых рефлексов из экспериментального набора данных (без заметного ущерба для полноты набора) может несколько снизить уровень шума функции трансляции [7].
После того как решения функции трансляции получены их уточняют с помощью процедуры оптимизации ориентации и положения модели как твердого тела по методу сопряженных градиентов (например, процедура FIT в AmoRe [11] или RIGID_BODY в CNS [6]).
2.1.1.3. Методы 6-мерного поискаПри использовании моделей плохого качества (например, в случае низкой гомологии) или моделей описывающих лишь малую часть неизвестной структуры часто возникает ряд проблем, затрудняющих решение задачи молекулярного замещения обычными методами. Значительные ошибки функции вращения, неизбежно возникающие в таких случаях, усугубляют собственные ошибки функции трансляции и приводят либо к полному отсутствию правильных решений, либо к тому, что эти решения оказываются среди максимумов, лежащих на уровне шума и нет достоверных критериев позволяющих однозначно выделить их среди прочих.
Единственным на сегодняшний день общим подходом, позволяющим решать вышеперечисленные проблемы и до определенной степени расширить границы применимости метода молекулярного замещения, является отказ от разделения задачи на поиск решений функций вращения и трансляции и применение процедуры 6-мерного поиска с одновременным варьированием как углов Эйлера (α,β,γ), так и компонент вектора трансляции (vx,vy,vz). Но, несмотря на значительный прогресс вычислительной техники, ни в одной из существующих программ, включая самые современные, 6-мерный поиск не проводится напрямую, как систематический поиск на 6-мерной сетке. Таким образом, ни одна из существующих программ не гарантирует нахождения абсолютных максимумов объединенной функции вращения-трансляции.
Не так давно, двумя группами независимо были предложены стохастические алгоритмы 6-мерного поиска, которые позволили создать программы, ставшие стандартным инструментом в рентгеновской кристаллографии макромолекул:
В работе Киссинджера и др. [26] был применен так называемый эволюционный алгоритм, который принадлежит к семейству алгоритмов стохастической оптимизации, включающему такие методы как Монте-Карло [28] и медленный отжиг [25].
Генетический алгоритм, независимо предложенный Чангом и Льюисом [14], основан на том же самом принципе, что и эволюционный и отличается от последнего лишь некоторыми деталями реализации. Подобный подход применялся также для разработки процедуры поиска положений тяжелых атомов в тяжелоатомных производных кристаллов макромолекул [13] и в прямых методах расчета фаз для кристаллов вирусных частиц [29].
Эволюционный алгоритм использует принцип естественного отбора для нахождения оптимальных решений. Вначале генерируется набор случайных решений, задающих одновременно и ориентацию и положение модели в элементарной ячейке. Затем рассчитываются структурные факторы Fm для каждого решения и производится отбор лучших решений исходя из коэффициента линейной корреляции [26].
Отобранные решения сохраняются и используются для создания нового набора с тем же количеством элементов, что и в предыдущем. Недостающие элементы нового набора получают, внося в ориентации и положения отобранных решений случайные изменения в соответствии с нормальным распределением. Таким образом, плотность распределения элементов нового набора уже не будет равномерной, а будет иметь максимумы в окрестностях отобранных решений. Затем снова происходит расчет структурных факторов Fm, отбор лучших решений, создание следующего набора, и так далее, пока не будет получено решение с некоторым оптимальным значением коэффициента линейной корреляции. На последней стадии, для лучшего отобранного решения проводится оптимизация ориентации и положения модели как твердого тела по методу сопряженных градиентов [33].
Скорость 6-мерного поиска с использованием эволюционного алгоритма значительно увеличивается за счет применения метода непрерывных преобразований структурных факторов [14, 26]. В этом методе структурные факторы рассчитываются один раз с помощью быстрого преобразования Фурье (FFT) для модели, помещенной в начало координат искусственной ячейки симметрии P1. В ходе 6-мерного поиска, изменение ориентации модели учитывается путем ортогональных преобразований индексов обратной решетки и использования линейной интерполяции в обратном пространстве. Изменение положения модели учитывается применением соответствующих фазовых сдвигов. При этом, принимаются во внимание вклады всех симметрически связанных молекул.
Одной из серьезных проблем, возникающих при уточнении фаз, полученных методом молекулярного замещения, является так называемая “model-bias” проблема [34]. Эта проблема проявляется в том, что если часть модели не соответствует реальной структуре, то на карте электронной плотности, рассчитанной по модели, данный участок будет в большей степени соответствовать модели чем реальной структуре. Традиционно, эта проблема решается с помощью OMIT карт электронной плотности, рассчитанных по модели, из которой исключен интересующий участок. С введением Брюнгером в программу CNS [6] возможности модификации электронной плотности рассчитанной по модели, появился еще один способ решения этой проблемы.
Таким образом, метод молекулярного замещения представляет собой мощный инструмент в кристаллографии макромолекул, который в случае высокой гомологии позволяет быстро и эффективно решать фазовую проблему. По мере того как количество известных структур макромолекул неуклонно растет, метод молекулярного замещения становится все более и более актуальным.
Похожие работы


... к родному языку автора, на этом языке (немецком) животное, из которого выделено вещество (лошадь), пишется как das Pferd. Белки и пептиды изучают чуть ли не во всех странах, а в научных публикациях на эту тему используются языки многих народов мира. Если в начале истории изучения этих веществ большинство научных работ считалось престижным писать на французском или немецком, то примерно с середины ...
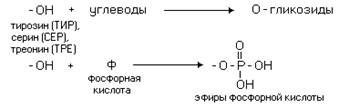
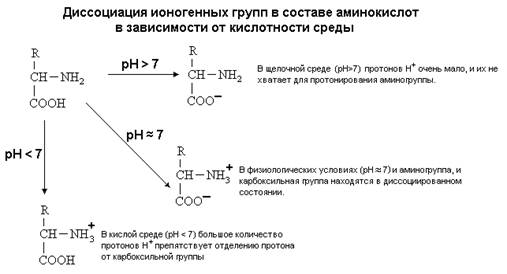
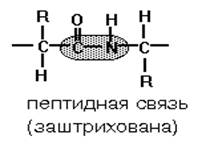
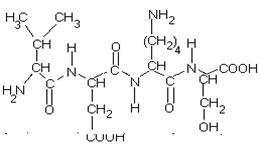
... равен 1/16 массы атома кислорода (кислородная единица массы). КОНФИГУРАЦИЯ И КОНФОРМАЦИЯ БЕЛКОВОЙ МОЛЕКУЛЫ Из всего сказанного можно заключить, что пространственная организация белков очень сложна. В химии существует понятие - пространственная КОНФИГУРАЦИЯ - жестко закрепленное ковалентными связями пространственное взаимное расположение частей молекулы (например: принадлежность к L-ряду ...
... , вызванные динамическими му-тациями.-----------------------T-----------T-------T-----T------T------T----------------------¬ Болезнь, номер по ¦ Ген, лока-¦Триплет¦Норма¦Прему-¦Мута- ¦Литература ¦ МакКьюсику (MIM) ¦ лизация ¦ ¦ ¦тация ¦ция ¦ ¦ -----------------------+-----------+-------+-----+------+------+----------------------+ Синдром ломкой X-хро- ¦FMR1, FRAXA¦(CGG)n ...
сте с этим закончился прежний, классический этап в развитии естествознания, характерный для эпохи Нового времени. Наступил новый этап неклассического естествознания XX века, характеризующийся, в частности, новыми, квантово-релятивистскими представлениями о физической реальности. 2. Научно-техническая революция и ее естественнонаучная составляющая Новые явления и процессы, имевшие место в ...
0 комментариев