Навигация
Обусловленные внутрисосудистым гемолизом;
28418
знаков
0
таблиц
0
изображений
1. Обусловленные внутрисосудистым гемолизом;
2. Обусловленные внесосудистым (внутриклеточным) гемолизом (эритроцитопатии; эритроэнзимопатии; гемоглобинопатии.)
Гемолитические анемии, обусловленные внутрисосудистым гемолизом. Возникают при токсических воздействиях (гемолитические яды, обширные ожоги), при инфекциях (сепсис, малярия), переливании несовместимой крови (посттрансфузионные).
Посттрансфузионные анемии по группе и резус-фактору крови делятся на изоиммунные и аутоиммунные. Изоиммунные анемии возникают при гемолитической болезни новорожденных: у матери (резус-отрицательной) вырабатываются антитела к эритроцитам плода. Возникает гемолитическая болезнь новорожденных, которая существует в трех формах: общий врожденный отек, врожденная анемия новорожденных, тяжелая желтуха новорожденных. Для общего врожденного отека (отечная форма анемии) характерны отек подкожной клетчатки, головного мозга и его оболочек, значительная гепато- и спленомегалия, гипертрофия миокарда. При врожденной анемии новорожденных (анемическая форма) находят малокровие внутренних органов и пневмонию. Тяжелая желтуха новорожденных развивается на вторые сутки, проявляется желтухой кожи и внутренних органов, а также области подкорковых ядер, печень и селезенка увеличены, в них находят эритробластоз и гемосидероз.
Аутоиммунные анемии появляются при группе ревматических болезней, медленных вирусных инфекциях, медикаментозных воздействиях, пароксизмальной холодовой гемоглобинурии.
Гемолитические анемии, обусловленные внесосудистым гемолизом. Для них характерен распад эритроцитов в макрофагах селезенки, костного мозга, лимфатических узлов, печени. В клинической картине типична триада: анемия, спленомегалия, желтуха. Выделяют три следующих вида.
Наследственные гемолитические анемии, связанные с нарушением мембраны эритроцитов — эритроцитопатии. При этом преобладают больные микросфероцитозом (в Европе — 200—300 на 1 млн человек); овалоцитоз и эллиптоцитоз встречаются реже.
Эритроэнзимопатии — нарушение активности ферментов эритроцитов. В патогенезе большую роль играет недостаток активности ферментов гликолиза и АТФ. Болезнь проявляется острыми гемолитическими кризами, реже течет как хроническая гемолитическая.
Гемоглобинопатии - в основе лежит нарушение синтеза гемоглобина. Lee и Cooly (1925) описали талассемию, в основе которой лежит нарушение синтеза одной из белковых цепей гемоглобина. Для нее характерен аутосомно-доминантный путь наследования. При талассемии резко уменьшается количество РНК, образуется фетальный гемоглобин. Заболевание проявляется выраженной или незначительной гипохромной анемией. Содержание железа сыворотки нормальное; развивается состояние гипоксии, бывают гемолитические кризы. Патология проявляется при инфекциях, медикаментозных воздействиях.
Выделяют также анемии, связанные с нарушением структуры цепей глобина Hb-S. B крови появляются аномальные гемоглобины. Тяжелая анемия возникает при гомозиготном состоянии, практически слабая — при гетерозиготном. При гемоглобине нарушается последовательность аминокислот в цепи — валин вместо глутамина. Эритроциты меняют свою форму, становятся серповидно-клеточными (серповидно-клеточная анемия) и быстро распадаются. Это наследственное заболевание более выражено в Африке, на Кубе, Ближнем Востоке, в Закавказье.
Тромбоцитарные заболевания.
Тромбоцитопатии — группа геморрагических заболеваний, связанных с врожденным или приобретенным снижением количества или изменением качества тромбоцитов.
Для клинико-морфологической картины тромбоцитопатии характерны множественные кровоизлияния, особенно петехиальные, и кровотечения различной локализации. Время кровотечения увеличено, ретракция сгустка снижена. Количество тромбоцитов в крови резко снижено или отмечаются их патологические формы.
Тромбоцитопения снижение количества тромбоцитов в 1единице объема крови.
Причины тромбоцитопении следующие.
I. Снижение продукции тромбоцитов
1. Приобретенное
Ø изменения костного мозга
Ø апластические и гипопластические анемии
Ø синдромы замещения костного мозга: лейкозные поражения и
Ø метастазы опухолей
Ø мегалобластные анемии
2. Специфически обусловленное
Ø лекарственные и токсические воздействия: алкоголь, цитоток-
сические препараты и др.
Ø вирусные инфекции — заболевания, вызванные вирусом Эпш-
тейна—Барр
3. Наследственное
Ø синдром Вискотта—Олдрича
II. Иммунологически опосредованное разрушение тромбоцитов
Ø Аутоиммунная тромбоцитопеническая пурпура
Ø Изоиммунные неонатальные и посттрансфузионные состояния Реакции лекарственной гиперчувствительности
Ø Вирусные инфекции (вирус Эпштейна—Барр, ВИЧ)
III. Увеличенная потребность в тромбоцитах
Ø ДВС-синдром
Ø Тромботическая тромбоцитопеническая пурпура
Ø Микроангиопатические нарушения: гемолитический уремический синдром
Ø Секвестрация (изоляция) тромбоцитов при гиперспленизме, гигантских гемангиомах
IV. Причины смешанного характера
Ø Тяжелые формы сепсиса
Ø Массивные гемотрансфузии.
Тромбоцитопения начинается когда количество кровяных пластинок в крови падает ниже 150х109/л..
Подавление тромбоцитопоэза иллюстрируется малым количеством мегакариоцитов в костном мозге и отмечается при апластических и гипопластических анемиях, мегалобластных анемиях и при замещении ткани костного мозга в ходе лейкоза или метастазировании опухоли с поражением костного мозга.
Иммунологически опосредованное разрушение тромбоцитов.
Аутоиммунная тромбоцитопеническая пурпура (Болезнь Верльгофа). Наблюдается в основном у детей и молодых людей.
У детей болезнь начинается остро, нередко после вирусной респираторной инфекции, продолжается в течение 2—4 нед. Тромбоциты разрушаются под действием антитромбоцитарных антител или иммунных комплексов. У молодых лиц (чаще 20—40-летних женщин) болезнь развивается постепенно и поддерживается в течение нескольких месяцев и даже лет. Обнаруживаются IgGS-антитела, которые дают перекрестные реакции с плацентой, поэтому встречаются тромбоцитопении неонатального периода.
Иногда аутоиммунная тромбоцитопения сочетается с с системной красной волчанкой, тяжелой миастенией, аутоиммунной гемолитической анемией. Она осложняет течение лимфом и лейкозов.
Кровоизлияния -от небольшой кожной пурпуры (множественных мелких кровоизлияний) до тяжелых внутри-маточных или желудочно-кишечных геморрагии. Встречаются кровоизлияния в головной мозг. Положительный лечебный эффект отмечается после иммуносупрессивной терапии и после спленэктомии, но уровень антител, связанных с тромбоцитами, не снижается.
Изоиммунная неонатальная и посттрансфузионная тромбоцитопении. Тромбоциты обладают специфическими изоантигенами. У 98 % взрослых людей имеется антиген Al (PLA-1), поэтому у PLA-1 -негативных лиц, получающих тромбоциты с этим антигеном при переливаниях крови, синтезируются соответствующие антитела. Во время беременности PLA-1 -отрицательная женщина может иммунизироваться по отношению к своему PLA-1-положительному плоду, при этом развивается неонатальная тромбоцитопения. Данное заболевание в сравнимо с изоиммунной гемолитической анемией.
Другие виды тромбоцитопении. Следует упомянуть о больных СПИДом, у которых тромбоцитопения —наиболее частое гематологическое проявление.
Тромбоцитарное истощение развивается при ДВС-синдроме и выраженном внутрисосудистом тромбозе. Кроме того, тромбоциты вместе с эритроцитами могут механически разрушаться в больших сосудистых опухолях, при микроангиопатической гемолитической анемии и тромботической тромбоцитопенической пурпуре.
Симптомы - ведущее значение имеют почечная недостаточность и кровотечения, спленомегалия.
Тромбоцитоз - увеличение числа тромбоцитов в периферической крови встречается при миелопролиферативных заболеваниях.
Вторичный тромбоцитоз развивается после спленэктомии, кровоизлияний, гемолиза, диссеминированном опухолевом процессе, хронических деструктивно-воспалительных заболеваниях (например, язвенном колите) и сверхсильной физической нагрузке.
Качественные аномалии тромбоцитов. Характеризуются нормальным количеством, но аномальной функцией тромбоцитов.
Бывают:
1. Наследственные
2. Приобретенные
Врожденные заболевания встречаются редко. В их основе лежат аутосомно-рецессивные нарушения синтеза мембранных гликопротеинов и секреции тромбоцитов.
К нарушению синтеза мембранных гликопротеинов относят:
1. Болезнь Гланцманна—Негели (тромбастения) – отсутствует агрегация у тромбоцитов, нарушенном связывании с фибриногеном, при продолжительных кровотечениях.
2. Синдром Бернара—Сулъе - обнаруживаются большие тромбоциты и снижение их способности к адгезии.
Нарушение секреции тромбоцитов
1. Болезнь фонда накопления – отсутствует электронно-плотных гранул в цитоплазме тромбоцитов и в нарушенном освобождении АДФ.
2. Недостаточность тромбоксансинтетазы, которая сопровождается нарушенным освобождением АДФ.
Приобретенные формы качественных аномалий тромбоцитов встречаются при уремии, печеночной недостаточности, миелопролиферативных заболеваниях и парапротеинемиях, при приеме больших доз аспирина и алкоголя. Аспирин необратимо блокирует циклооксигеназу тромбоцитов, что снижает синтез простагландинов и тромбоксана А2.
Геморрагические диатезы (коагулопатии).
Расстройства коагуляции могут иметь приобретенную и наследственную природу.
![]() Приобретенные коагулопатии включают аномалии свертывания крови.
Приобретенные коагулопатии включают аномалии свертывания крови.
Ø Недостаточность витамина К приводит к подавлению синтеза факторов коагуляции II, VII, IX, X и белка С.
Ø В печени продуцируются практически все факторы коагуляции, тяжелые ее поражения вызвают геморрагический диатез.
Ø ДВС-синдром ведет к недостаточности факторов коагуляции.
Наследственные коагулопатии
Из истории известно, что отдельные королевкие династии Европы страдали разными формами гемофилии, которая относится к данной группе. Возникшие очень давно в результате браков между родственниками, они передавались по наследству потомкам в другим монаршим домам (например русский цесаревич Алексей, сын императора Николая Второго и немецкой принцессы Александры).
Недостаточность фактора VIII (гемофилия А) и фактора IX (гемофилия В) передается как рецессивное заболевание, связанное с половыми хромосомами.Для других наследственных заболеваний характерен аутосомный тип.
Природу нарушения гемостаза можно распознать с помощью учета 4 параметров:
1. Времени кровотечения
2. Количества тромбоцитов в крови,
3. Протромбинового времени (продолжительность формирования свертка плазмы крови в присутствии тромбопластина и солей кальция в секундах)
4. Тромбопластинового времени (периода формирования тромбопластина, способствующего превращению протромбина в тромбин).
На основе этих данных выделяют 4 вида коагулопатии.
Недостаточность комплекса фактор VIII — фактор Вилле-бранда (VIII — vWF).
Дефекты комплекса VIII — vWF, имеющие генетическую природу, вызывают два наследственных заболевания с геморрагическим диатезом — гемофилию А и болезнь Виллебранда.
Болезнь Виллебранда (vWD) - частота около 1 % среди наследственных геморрагических диатезов.
Характеризуется спонтанными кровотечениями из слизистых оболочек внутренних органов, избыточными кровотечениями из ран и при менорагиях, увеличенным временем кровотечения при нормальном количестве тромбоцитов в крови.
Передается по аутосомно-доминантному типу, однако известны и редкие аутосомно-рецессивные варианты. Описано более 20 форм vWD, которые разделены на две главные группы.
В 1-ю группа - объединяет I и III типы, ко 2 группе относят II тип заболевания.
Типы I и III - связаны с уменьшенным количеством фактора Виллебранда (vWF). 70 % всех наблюдений относятся к типу I с легким течением и передающимся по аутосомно-доминантному типу.
III тип - аутосомно-рецессивный, связан с крайне низкими уровнями vWF и более тяжелым течением болезни. Он встречается реже, чем тип I.
Тип II - характеризуется качественным дефектом фактора Виллебранда и наследуется по аутосомно-доминантному типу, формируется аномальный vWF. При этом объемы кровопотери варьируют от небольших до умеренных. У людей с болезнью Виллебранда имеется сложное нарушение функций тромбоцитов и системы коагуляции крови.
Гемофилия А (недостаточность фактора VIII). Самая часто встречаемая в данной группе, протекает с сильными кровотечениями. Развивается из-за уменьшения количества или активности фактора VIII. Он служит кофактором для активации фактора X.
Наследуется как Х-связанный рецессивный признак и поэтому встречается у мужчин и гомозиготных женщин. Вместе с тем и у гетерозиготных женщин описаны избыточно обильные кровотечения. Около 30 % больных не имеют ни одного родственника с подобной патологией, возможно что заболевание у них возникло в результате мутаций.
Проявления гемофилии А зависят от активности фактора VIII. При активности менее 1 % нормы проявляется тяжелая форма, 2—5 % — умеренно тяжелая, 6—50 % — легкие формы гемофилии А.
Во всех случаях характерны:
1. Тенденция к массивным кровоизлияниям после травм или хирургических вмешательств.
2. Спонтанные кровотечения в крупные суставы, несущие наибольшую механическую нагрузку, приводят к гемартрозам без видимой травмы.
3. Повторные гемартрозы заканчиваются деформациями и ин-валидизацией суставов.
4. Ни петехии, ни экхимозы нехарактерны.
5. Время кровотечения и количество тромбоцитов нормальные,
6. Увеличено тромбопластиновое время.
7. Для диагностики нужна оценка содержания фактора VIII.
Гемофилия В (болезнь Кристмаса, недостаточность фактора IX). Тяжелая степень недостаточности фактора IX представляет собой заболевание, клинически не отличимое от гемофилии А. Наследуется как Х-связанный рецессивный признак и может протекать бессимптомно или с кровоизлияниями. Примерно у 14 % больных фактор IX обнаруживается, но в нефункциональном состоянии. Как и при гемофилии А, время кровотечения нормально, а неполное тромбопластиновое время увеличено.
Похожие работы
... сделать выводы. После проведения курса лечения препаратами железа рекомендуют для закрепления эффекта повторять курсы 2-3 раза через полгода. Таким образом, весь процесс лечения анемии составляет около 2 лет. ПРЕПАРАТЫ ЖЕЛЕЗА. Все препараты содержат несколько видов солей железа. Чаще всего сульфат железа (феррокаль, тардиферон,ферроплекс и т.д.); аскорбинат железа, лактат железа, трехвалентное ...
... и одобряемых целей и средств, желая создать новую систему норм и ценностей, а так же средств их достижения. В типологии социальных отклонений выделяются и такие типы девиантного поведения, как культурные и психологические отклонения, индивидуальные и групповые отклонения, первичные и вторичные отклонения, культурно одобряемые отклонения, культурно осуждаемые отклонения. Девиация может носить ...
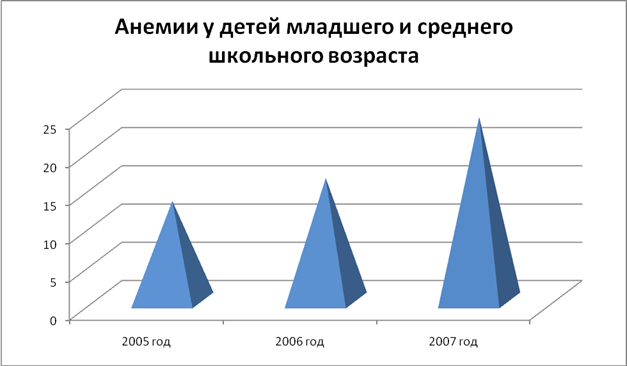
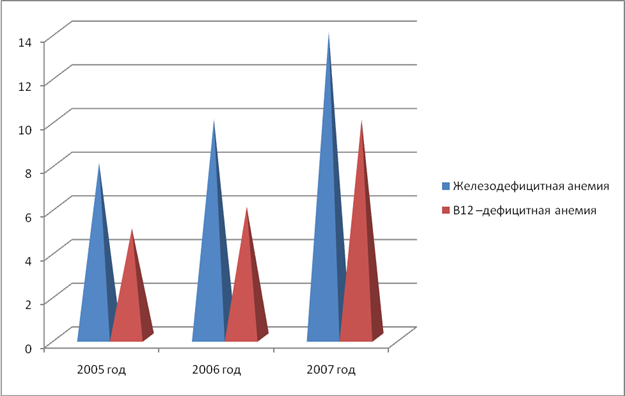
... 1 Статистические данные заболеваемостью анемиями по Касимову и Касимовскому району среди детей за 2005 -2007 гг 2005 год 2006 год 2007 год Анемии у детей младшего и среднего школьного возраста 13 16 24 Диаграмма 1 Таблица 2 Соотношение заболеваемости железодефицитной В12-дефицитной анемиями среди детей за 2005 – 2007 гг. 2005 год 2006 ...
... (лизоцим, пропердин и т.д.), так и специфический иммунитет. Показано, что в отсутствие железа IgA теряет свою бактерицидную активность. - Сердечнососудистый синдром наблюдают при тяжёлой железодефицитной анемии. У детей развиваются повышенная утомляемость, низкое артериальное давление, тахикардия, снижение тонуса сердечной мышцы, приглушение тонов, функциональный, довольно грубый ...
0 комментариев