Навигация
Способы получения синтез-газа
100076
знаков
5
таблиц
23
изображения
2. Способы получения синтез-газа
Первым способом получения синтез-газа была газификация каменного угля, которая была осуществлена еще в 30-е годы XIX века в Англии с целью получения горючих газов: водорода, метана, монооксида углерода. Этот процесс широко использовался во многих странах до середины 50-х годов XX века, а затем был вытеснен методами, основанными на использовании природного газа и нефти. Однако в связи с сокращением нефтяных ресурсов значение процесса газификации снова стало возрастать.
В настоящее время существуют три основных промышленных метода получения синтез-газа[34].
1. Газификация угля. Процесс основан на взаимодействии угля с водяным паром:
C + H2O ↔ H2 + CO
Эта реакция является эндотермической, равновесие сдвигается вправо при температурах 900-1000 оС. Разработаны технологические процессы, использующие парокислородное дутье, при котором наряду с упомянутой реакцией протекает экзотермическая реакция сгорания угля, обеспечивающая нужный тепловой баланс:
C + 1/2O2↔CO
2. Конверсия метана. Реакция взаимодействия метана с водяным паром проводится в присутствии никелевых катализаторов (Ni-Al2O3) при повышенных температурах (800-900 оС) и давлении:
CH4 + H2O → CO + 3H2
В качестве сырья вместо метана может быть использовано любое углеводородное сырье.
3. Парциальное окисление углеводородов. Процесс заключается в неполном термическом окислении углеводородов при температурах выше 1300 оС:
CnH2n+ 2 + 1/2nO2 → nCO + (n + 1)H2
Способ применим к любому углеводородному сырью, но наиболее часто в промышленности используют высококипящую фракцию нефти - мазут.
Соотношение СО : Н2 существенно зависит от применяемого способа получения синтез-газа. При газификации угля и парциальном окислении это соотношение близко к 1 : 1, тогда как при конверсии метана соотношение СО : Н2 составляет 1 : 3. В настоящее время разрабатываются проекты подземной газификации, то есть газификации угля непосредственно в пласте. Интересно, что эта идея была высказана Д.И. Менделеевым более 100 лет назад. В перспективе синтез-газ будут получать газификацией не только угля, но и других источников углерода вплоть до городских и сельскохозяйственных отходов.
3. Газификация угля
Газификация — высокотемпературный процесс взаимодействия углерода топлива с окислителями, проводимый с целью получения горючих газов (Н2, СО, СН4). В качестве окислителей, которые иногда называют газифицирующими агентами, используют кислород (или обогащенный им воздух), водяной пар, диоксид углерода либо смеси указанных веществ. В зависимости от соотношения исходных реагентов, температуры, продолжительности реакции и других факторов можно получать газовые смеси самого разного состава.
3.1 Тенденции развития и новые инженерные решения в газификации угляВпервые промышленная реализация газификации твердых топлив была осуществлена в 1835 г в Великобритании. К 50-м годам XIX в. практически во всех крупных и средних городах Европы и Северной Америки действовали газовые заводы для производства отопительного, бытового и светильного газа [4]. К середине XX в этот процесс получил широкое развитие в большинстве промышленных стран мира. Например, в СССР в 50-е годы работало свыше 350 газогенераторных станций, на которых было установлено около 2500 газогенераторов. Эти станции вырабатывали ежегодно 35 млрд. м3 энергетических и технологических газов. Это был "золотой век" газификации угля. Начиная с 60-х годов XIX в., все более серьезную конкуренцию углю начинает оказывать нефть. В начале 1960-х годов разработка месторождений дешевой нефти на Ближнем Востоке и в Западной Сибири привела практически к полной ликвидации этой отрасли промышленности. Как известно, в последующие 20—25 лет в мировом энергетическом балансе происходили изменения, обусловленные ростом добычи и потребления нефти, попутных и природных газов. Вследствие этого конкурентоспособность искусственных энергетических и технологических газов, получаемых из твердых топлив, резко снизилась, и их производство практически повсеместно было прекращено. Сохранились лишь небольшие островки в уникальных регионах. Например, в ЮАР углепереработка (главным образом на основе газификации угля) стала крупной промышленным сектором из-за эмбарго на поставку нефти. Началось триумфальное шествие нефти. Однако уже в 1972 г. оно омрачилось первым "энергетическим кризисом", который по существу был спровоцирован на политической основе странами-участниками ОПЕК. Мировые цены на нефть подскочили с 5-7 до 24 долл. США за баррель (1 т сырой нефти сорта Brent ≈ 8,06 баррелей), и стало ясно, что углепереработку списывать в архив рано, так как в большинстве развитых стран много угля и мало или совсем нет нефти.
Этот кризис преподнес цивилизованному миру очень важный урок. Во-первых, все осознали, что запасы углеводородного сырья распределены крайне неравномерно и неудобно, и, во-вторых, эти запасы - исчерпаемы. Запасы же угля и других твердых горючих ископаемых – нефтяных сланцев, битумных песков, торфа и т.п. распределены более равномерно, и сроки их исчерпания оценивается многими сотнями лет. Но самый главный результат этот кризиса заключается в активизации работ по энергосбережению.
Однако в последние годы в связи с сокращением ресурсов нефтяного и газового сырья процесс газификации твердых горючих ископаемых вновь привлек к себе внимание, искусственные газы опять начинают рассматриваться как одна из существенных составляющих теплового баланса. Например, в США планировалось к 1990 г построить 63 завода этого профиля средней мощностью ~7 млн. м3 газа в сутки каждый. Их годовая выработка составляла 140 млрд. м3, а к 2000 г увеличилась до 220—250 млрд. м3, что соответствует ~23% потребности США в энергетических и технологических газах.
3.2 Взгляд на углепереработку сквозь десятилетияВ середине 1980-х годов интерес к углепереработке пошел на убыль. Причин несколько.
Во-первых, политикой "кнута и пряника" США установили контроль над странами - производителями нефти. Наиболее амбициозных (Ирак, Иран) наказали в назидание другим. В результате рост цен на нефть замедлился. Сохранять равновесие поручили шестому флоту США и силам быстрого реагирования. Насколько это равновесие устойчиво покажет время.
В течение 1980-х годов цены на нефть снизились с 40 долл. США за баррель (что соответствует примерно 65 долл. США за баррель в современных ценах с поправкой на инфляцию) до минимального уровня 9,13 долл. США за баррель в декабре 1998 г. и в настоящее время колеблются в "коридоре" 17-27 долл. США за баррель.
Во-вторых, эффективно сработали государственные программы энергосбережения, что в конечном итоге привело к снижению темпа роста потребления нефти и природного газа. С середины 1970-х годов энергоемкость единицы ВВП в развитых странах снизилась на 22 %, а нефтеемкость на 38 % [5].
В-третьих, динамичное развитие нефтегазовой отрасли и масштабные работы по разведке новых месторождений нефти и газа показали, что запасы углеводородного сырья на самом деле значительно больше, чем предполагалось. Последние 20 лет ежегодный прирост разведанных запасов нефти и газа опережает их потребление, и прогнозные сроки исчерпания регулярно отодвигаются. По достаточно авторитетным данным глобальную замену нефти углем следует ожидать после середины XXI в., а замену природного газа углем – к концу века. Если, конечно, не произойдет прорыва в развитии технологии ядерного синтеза.
В-четвертых, ни одна из разрабатываемых технологий не позволила повысить рентабельность процесса получения жидкого топлива из угля в такой степени, чтобы "синтетическая нефть" могла конкурировать с природной нефтью.
В итоге “эпоха угля” не наступила и интерес к переработке угля уменьшился. Большинство программ было свернуто, а оставшиеся - радикально урезаны. Более десятка проектов были завершены на стадии 5-летней готовности, т.е. при изменении конъюнктуры рынка углеводородного сырья можно в течение 5 лет на основе демонстрационных установок производительностью 10-60 т/ч по углю развернуть промышленное производство. Если от коммерческого использования технологий прямого и непрямого ожижения угля в конце 1980-х гг. пока отказались, то интерес к газификации угля хотя и уменьшился, но не прекратился. Например, в ряде регионов, где природного газа нет или мало (Северная Америка, Китай и др.), использование газа из угля для синтеза метанола и аммиака экономически оправдано и построен ряд промышленных предприятий.
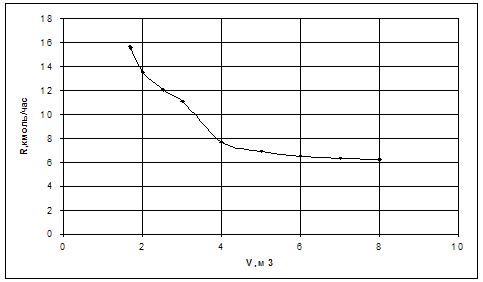
В 1990-е годы бурное развитие получила внутрицикловая газификация для производства электроэнергии, т.е. использование бинарного цикла, при котором горючий газ утилизируется в газовой турбине, а продукты сгорания используются при генерации пара для паровой турбины. Первая коммерческая электростанция с внутрицикловой газификацией – Cool Water, США, шт. Калифорния, мощностью 100 МВт (60 т/ч по углю) была построена в 1983 г. Использовался газогенератор Texaco с подачей топлива в виде водо-угольной суспензии. После 1993 г. в разных странах было введено в эксплуатацию 18 электростанций с внутри цикловой газификацией твердого топлива мощностью от 60 до 300 МВт. На рис.1 приведены данные по мировому производству газа из твердых топлив с 1970 г., а в табл. 1 – структура его потребления.
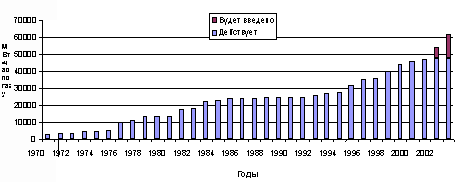
Рис. 1. Суммарная мощность газогенераторных установок
Таблица 1
Динамика потребления газа из угля в мире| Целевое использование | Использование в 2001 г., МВт по газу | Доля в 2001 г., % | Вводится в эксплуатацию до конца 2004 г., МВт по газу | Годовой прирост мощности в 2002-2004 гг., % |
| Химическое производство | 18 000 | 45 | 5 000 | 9,3 |
| Внутрицикловая газификация (производство электроэнергии) | 12 000 | 30 | 11 200 | 31 |
| Синтез по Фишеру-Тропшу | 10 000 | 25 | 0 | 0 |
| ВСЕГО | 40 000 | 100 | 17 200 | 14,3 |
Приведенные данные наглядно демонстрируют ускорение динамики вовлечения газификации угля в мировую промышленность. Повышенный интерес к внутрицикловой газификации угля в развитых странах объясняется двумя причинами.
Во-первых, ТЭС с внутрицикловой газификацией экологически менее опасна. Благодаря предварительной очистке газа сокращаются выбросы оксидов серы, азота и твердых частиц.
Во-вторых, использование бинарного цикла позволяет существенно увеличить КПД электростанции и, следовательно, сократить удельный расход топлива.
В табл. 2 приведены характерные величины удельных выбросов и КПД для ТЭС с внутрицикловой газификацией и для ТЭС с традиционным сжиганием угля.
Таблица 2
Величины удельных выбросов и КПД для ТЭС с внутрицикловой газификацией и с традиционным сжиганием угля
| Параметры | Традиционная угольная ТЭС | ТЭС с внутрицикловой газификацией |
| Концентрация вредных веществ в дымовых газах | 130 | 10 |
| Электрический КПД, % | 33-35 | 42-46 |
Необходимо отметить, что удельные капитальные затраты при использовании внутрицикловой газификации составляют примерно 1500 долл. США за 1кВт с перспективой снижения до 1000-1200 долл. США, в то время как для традиционной угольной ТЭС удельные капитальные затраты составляют примерно 800-900 долл. США за 1 кВт. Ясно, что ТЭС с внутрицикловой газификацией твердого топлива более привлекательна при наличии экологических ограничений в месте размещения и при использовании достаточно дорогого топлива, так как расход топлива на 1 кВт сокращается.
Эти условия характерны для развитых стран. В настоящее время использование внутрицикловой газификации твердого топлива считается самым перспективным направлением в энергетике.
3.3 Инженерные разработки за прошедшее столетие
В настоящее время выявились следующие наиболее экономически эффективные области применения метода газификации:
- газификация сернистых и многозольных топлив с последующим сжиганием полученных газов на мощных тепловых электростанциях. В углях, ежегодно добываемых в России, содержится около 10 млн. т серы, большая часть которой при сжигании выбрасывается в атмосферу в виде токсичных оксидов серы и серооксида углерода. При газификации сернистых углей образуется сероводород, который можно сравнительно легко извлечь и затем переработать в товарную серу или серную кислоту
- газификация твердых топлив для крупномасштабного производства заменителей природного газа. Это направление имеет наибольшее значение для местного газоснабжения районов, удаленных от месторождений природного газа и нефти или от магистральных трубопроводов
- газификация твердых топлив с целью получения синтез-газа, газов-восстановителей и водорода для нужд химической, нефтехимической и металлургической промышленности.
Процесс газификации зависит от многих факторов, влияющих на состав получаемого газа и его теплоту сгорания. В связи с этим до сих пор отсутствует единая общепринятая классификация методов осуществления рассматриваемого процесса. Ниже приведен один из возможных вариантов классификации.
· по виду дутья (газифицирующего агента): воздушное, воздушно-кислородное, паровоздушное, парокислородное.
· по давлению: при атмосферном давлении, при повышенном давлении.
· по размеру частиц топлива: газификация крупнозернистого (кускового), мелкозернистого и пылевидного топлива.
· по конструктивным особенностям реакционной зоны: в неподвижном плотном слое топлива, в псевдоожиженном слое топлива, в пылеугольном факеле.
· по способу выведения золы: в твердом виде, в виде жидкого шлака.
· по способу подвода тепла: при частичном сжигании топлива в газогенераторе, при смешении топлива с предварительно нагретым твердым, жидким или газообразным теплоносителем (регенеративный нагрев), при подводе тепла через стенку аппарата (рекуперативный нагрев).
· по назначению получаемого газа: получение газов с заданной теплотой сгорания (низкой — до 6700 кДж/м3, средней — от 12000 до 18000 кДж/м3 и высокой — от 30000 до 35000 кДж/м3); получение газов заданного состава.
· по способу обогащения конечного газа метаном: безостаточная газификация топлива в СО, СО2 и Н2 в сочетании с отдельной стадией метанирования СО и СО2 водородом; газификация с полным выделением летучих и максимальным образованием метана в слое топлива; гидрогазификация.
Газификации может быть подвергнуто большинство известных видов твердых горючих ископаемых. При этом можно получить газ заданного состава или заданной теплоты сгорания, так как эти показатели в значительной степени определяются температурой, давлением и составом применяемого дутья.
Газ с низкой теплотой сгорания образуется при использовании воздушного или паровоздушного дутья. В соответствии с этим его называют воздушным или паровоздушным (смешанным). Он характеризуется высоким содержанием балласта — азота (до 40—50% об.), что обусловливает низкую теплоту сгорания такого газа. Основная область применения таких газов — сжигание в топках промышленных печей. Кроме того, после, конверсии содержащегося в них оксида углерода и очистки от СО2 получают азотоводородную смесь — исходное сырье для синтеза аммиака.
Газы со средней теплотой сгорания получают в процессах паровой или парокислородной газификации твердых топлив под давлением до 2—2,5 МПа. По составу они представляют собой смеси оксидов углерода и водорода с небольшими количествами метана и других углеводородов: 30—35% (об.) СО2, 10—13% (об.) СО, 38—40% (об.) Н2) 10—12% (об.) СН4, 0,5— 1,5% (об.) СnН2n. По экономическим соображениям такие газы применяют в ограниченных масштабах. Их используют главным образом как химическое сырье, а также начинают применять в металлургии в качестве газов-восстановителей.
Технология получения указанных газов первоначально была основана на использовании паровоздушного дутья, причем воздух предварительно обогащался кислородом до 40% (об.). Наряду с этим повысить теплоту сгорания газа можно, проводя газификацию при повышенном давлении. Другой способ получения газов со средней теплотой сгорания — газификация твердых топлив с применением парового дутья и предварительно нагретого до 900—1100 °С твердого теплоносителя. В качестве последнего можно использовать золу, остающуюся после сжигания части топлива в выносной топке. Подобный вариант позволяет получать газ, состоящий в основном из СО и Н2 в соотношении, близком к 1:1, однако этот способ опробован пока лишь на небольших опытно-промышленных установках.
Газы с высокой теплотой сгорания, приближающиеся по этому показателю к природному газу, в настоящее время в промышленных масштабах пока не производят. Однако технология их получения в ряде случаев отработана на достаточно крупных опытно-промышленных установках. Основа повышения теплоты сгорания газа — обогащение его метаном за счет проведения газификации при повышенном давлении, благодаря чему интенсифицируется взаимодействие углерода и его оксидов с водородом, образующимся в слое топлива. Продуктом этих реакций является метан. [7]
Разработано также несколько вариантов многоступенчатых газогенераторов, в которых предусмотрены максимальное извлечение летучих продуктов из топлива и последующая газификация углеродного остатка с применением водородсодержащих газов в качестве газифицирующего агента (гидрогазификация). Наряду с этим газ, обогащенный метаном, может быть получен из низко- и среднекалорийного газа путем гидрирования содержащихся в нем оксидов углерода в выносном реакторе (вне газогенератора).
Для современной химической промышленности и энергетики требуются газогенераторы с единичной мощностью по углю 100 т/ч и более. К началу 1970-х годов в промышленном масштабе было реализовано три типа газогенераторов [6].
· слоевые газогенераторы. В разное время действовало более 800 газогенераторов, в том числе более 30 газогенераторов “Лурги” с единичной мощностью по углю до 45 т/ч. После 1977 г. введено в эксплуатацию еще 130 газогенераторов “Лурги”.
· газогенераторы Винклера с кипящим слоем. Было сооружено более 40 аппаратов с единичной мощностью до 35 т/ч по углю.
· пылеугольные газогенераторы Копперса-Тотцека. К началу 1970-х годов эксплуатировалось более 50 аппаратов с единичной мощностью до 28 т/час по углю.
Не случайно все самые мощные газогенераторы имели немецкое происхождение. Причина в том, что в Германии нет собственной нефти, но имеются большие запасы угля. В 1920-1940 гг. в Германии была реализована беспрецедентная по масштабам программа углепереработки с производством моторных топлив, металлургического топлива, газов различного назначения и широкого спектра продуктов углехимии, включая пищевые продукты. Во время второй мировой войны с использованием жидких продуктов пиролиза, прямого и непрямого ожижения угля производилось до 5,5 млн. т в год моторного топлива. Именно немецкие разработки того времени определили на многие десятилетия стратегию развития технологий углепереработки, в том числе газификации топлива.
Если проанализировать конструктивные особенности и принцип действия современных промышленных газогенераторов (к настоящему времени до промышленного масштаба доведено еще более десяти конструкций газогенераторов), можно выделить четыре основополагающих инженерных решения.
1. Создание Фрицем Винклером (концерн BASF) в 1926 г. газогенератора с кипящим слоем. Эта технология послужила основой для современных процессов HTW (Hoch-Temperatur Winkler) и KRW (Kellogg-Rust-Westinghouse) и др.
2. Разработка фирмой "Лурги" в 1932 г. слоевого газогенератора, работающего под давлением 3 МПа. Использование повышенного давления для интенсификации процесса газификации реализовано почти во всех современных промышленных газогенераторах.
3. Разработка Генрихом Копперсом и Фридрихом Тотцеком в 1944-45 гг. пылеугольного газогенератора с жидким шлакоудалением. Первый промышленный аппарат этого типа был построен в 1952 г. в Финляндии. Пылеугольный принцип газификации с жидким шлакоудалением реализован в промышленных аппаратах Destec, Shell, Prenflo, разработанных на основе газогенератора Копперса-Тотцека, в аппарате Texaco и др. Удаление шлака в жидком виде реализовано в слоевом газогенераторе BGL (British Gas– Lurgy), разработанном на основе газогенератора Лурги.
4. Разработка фирмой Texaco в 1950-е годы газификаторов для переработки тяжелых нефтяных остатков. Всего построено более 160 таких установок. В 1970-е годы была разработана модификация аппарата Texaco для газификации водо-угольной суспензии. Принцип подачи угля в аппарат в виде водо-угольной суспензии использован и в газогенераторе Destec.
Были попытки использовать и ряд других технических решений для создания новых газогенераторов: использование внешнего теплоносителя, в том числе тепла ядерного реактора; газификация в расплавах солей, железа, шлака; двух - трехступенчатая газификация; газификация в плазме; каталитическая газификация и др., но они не привели к созданию современного конкурентоспособного технологического процесса.
4. Конверсия метана в синтез-газ
Углекислотная конверсия метана в синтез-газ СО + Н2 - одна из важнейших химических реакций, пригодная для промышленного получения водорода и дающая начало синтезу углеводородов (жидкое топливо) и других технически ценных продуктов.
Существует три метода окислительной конверсии метана в синтез-газ:
· паровая конверсия
CH4 + H2O ↔ CO + 3H2
ΔН = +206 кДж/моль (1)
· парциальное окисление кислородом
CH4 + 1/2O2 ↔ CO + 2H2
ΔН = -35,6 кДж/моль (2)
· углекислотная конверсия
2CO + 2H2 ↔ CH4 + CO2
ΔН = +247 кДж/моль (3)
В промышленности используется практически лишь метод паровой конверсии (1). Реакцию проводят на нанесенном Ni-катализаторе при высокой температуре (700 - 900 °С). Что касается реакции (2), то на ее основе фирмой «Shell» был разработан технологический процесс в некаталитическом варианте при очень высоких температурах (1100 - 1300 °С), реализованный на небольшом заводе в Малайзии. Заметим, что по последним сведениям из-за аварии этот завод сейчас не работает. Реакция (3) пока находится в стадии исследования на уровне лабораторных и пилотных испытаний. Как следует из уравнений (1) - (3), количественный состав образующегося синтез-газа в этих реакциях различный: в реакции (1) получается синтез-газ состава СО:Н2 = 1:3, в реакции (2) - смесь 1:2, в реакции (3) - смесь 1:1. Потребность в синтез-газе того или иного состава определяется его последующим техническим назначением.
Так, для синтеза метанола требуется синтез-газ состава 1:2
СО + 2Н2 = СН3ОН (4)
В производстве аммиака из азото-водородной смеси на стадии ее получения применяют синтез-газ состава 1CO:3H2. Относительно недавно предложено использовать синтез-газ состава 1:1 для промышленного получения диметилового эфира [9, 10]. Формальная стехиометрия этой реакции соответствует уравнению
2СО + 4Н2 = СН3ОСН3 + Н2О (5)
Однако, с учетом того, что в условиях этого процесса H2O вступает во взаимодействие с CO (паровая конверсия CO)
CO + H2O ↔ CO2 + H2
ΔH = - 41 кДж/моль (6)
реально для получения диметилового эфира требуется смесь CO:H2 состава 1:1:
3СО + 3Н2 = СН3ОСН3 + СО2 (7)
Термодинамическое рассмотрение реакции (7) указывает, что она может осуществляться при давлениях значительно меньших, чем реакция (4). Катализатором реакции (7) может служить комбинация катализаторов дегидратации и синтеза метанола. Получаемый диметиловый эфир предлагается применять в качестве топлива в дизельных двигателях без переделки самих двигателей (это топливо резко снижает вредные выхлопы ─ «топливо 21 века», как его назвали разработчики).
4.1 Термодинамика процесса Равновесие в системе 2CO + 2H2 ↔ CH4 + CO2Большие трудности в практическом осуществлении всех методов конверсии метана связаны со значительным тепловым эффектом: как эндотермичность реакций (1) и (3), так и экзотермичность реакции (2) создают проблему подвода или отвода тепла. В углекислотной конверсии метана (3) при 700 - 800 °С на многих никелевых и платиновых катализаторах достигается равновесная конверсия в синтез-газ СО + Н2. В этих условиях одновременно с реакцией (3) осуществляется взаимодействие монооксида углерода с водяным паром (6). Протекание реакции (6) приводит к тому, что в равновесии (3) отношение СО:Н2 оказывается меньше 1, а конверсия СО2 больше конверсии СН4. Лишь при 900 °С и атмосферном давлении выход Н2 и СО приближается к 100%, а отношение Н2О/СО к нулю.
На рис. 11 показана зависимость равновесного выхода Н2 и СО в исходной системе CH4 + CO2 от температуры и давления.
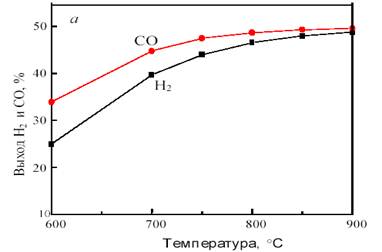
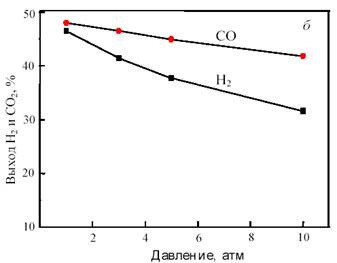
Рис. 11. Зависимость равновесного выхода Н2 и СО от температуры при 0,1 МПа (а) и от давления при 800 °С (б) в исходной смеси 1СН4:1СО2
Как видно из рис. 11, с повышением температуры выход водорода и CO возрастает, достигая предела вблизи 900 °С. С ростом давления равновесная конверсия уменьшается.
Основным препятствием к использованию Ni-катализаторов отравляемость коксом. Возможны два пути образования кокса при разложении метана:
· диссоциация метана
СН4 = С + 2Н2
ΔН = +74,8 кДж/моль•С (8)
· реакция Будуара
2CO ↔ C + CO2
ΔН = -172,5 кДж/моль•С (9)
Первая из них ─ эндотермическая, вторая ─ экзотермическая. Обе реакции могут быть представлены как стадии суммарной реакции (3). Однако в реальности они протекают при разных температурах: реакция (8) ─ преимущественно при высоких температурах, реакция (9) ─ при низких температурах, и в реальных условиях кокс почти всегда образуется. Согласно термодинамическим соображениям суммарное углеотложение должно снижаться с повышением температуры. Действительно, эксперимент подтверждает, что основное количество углерода образуется по реакции (8), а не (9). Часто углерод, диффундируя в металл, образуется на выходе из катализатора в виде нитей.
Одним из путей решения проблемы, связанной с подводом и отводом тепла при получении синтез-газа, является разработка процесса комбинированной конверсии смеси СН4 + СО2 + Н2О + О2, в котором бы без дополнительного подогрева сочетались реакции (1), (2), (3) и (6). Такую термонейтральную (автотермическую) конверсию можно осуществить, комбинируя углекислотную (3) и кислородную (2) конверсию метана в системе СН4 + СО2 + О2. Термодинамический расчет процесса комбинированной конверсии, включающей реакции (2), (3) и паровой конверсии СО (6), показывает, что в смеси 50% СН4 + (50 - х)% СО2 + х% О2 при 800 °С термонейтральность достигается при х = 23% (рис. 12). В реакции смеси 50% СН4 + 27% СО2 + 23% О2 при 800 °С и 1 атм. равновесные выходы составляют: 49,3% Н2 и 36,5% СО, т.е. соотношение CO:H2 сильно отличается от единицы.
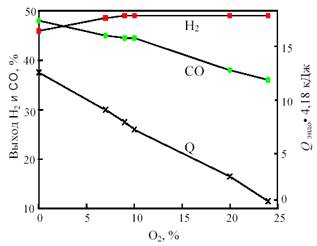
Рис. 12. Зависимость равновесного выхода Н2 и СО и теплового эффекта реакции (теплопоглощения) Qэндо при 800 °С и 0,1 МПа от содержания кислорода (x) в смеси 50% СН4 + (50-х)% СО2 + х% О2
Изменение соотношения исходных компонентов позволяет получить газ состава 1СО:1Н2 с одновременным сохранением термонейтральности. Например, исходная смесь, содержащая 38% СН4, 43% СО2 и 19% СО2, при 800 °С и 1 атм. дает продукт состава 36,0% Н2 и 36,4% СО при нулевом тепловом эффекте. При повышении температуры получается избыток СО: при 900 °С ─ 34,6% Н2 и 38,0% СО, а при снижении температуры ─ избыток Н2: при 700 °С ─ 36,4% Н2 и 33,6% СО. В качестве примера на рис. 13 показана зависимость равновесного выхода СО и Н2 от температуры и давления для исходной смеси 38 % CH4 + 43% CO2 + 19% O2.
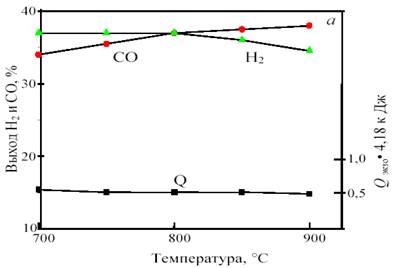
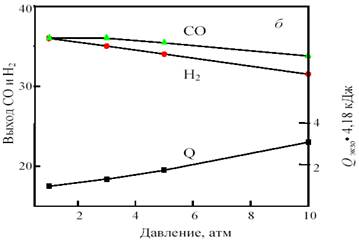
Рис. 13. Зависимость равновесного выхода Н2 и СО и теплового эффекта реакции (тепловыделения) Qэндо при 0,1 МПа от температуры (а) и при 800 °С от давления (б) в смеси 38%СН4 +43%CO2 + 19%O2
Важно отметить, что для этой смеси, в отличие от смеси 1СО:1Н2, с ростом давления от 1 до 10 атм. равновесный выход продуктов уменьшается не намного, всего на 2─3%. Это позволяет интенсифицировать процесс путем увеличения давления без изменения соотношения продуктов и термонейтральности.
Похожие работы
... эту высокую стоимость. К тому же метанол сильно ядовит. Что касается дизельных двигателей, то в них можно использовать продукт разложения метанола – ДМЭ. 4. Получение диметилового эфира дегидратацией метанола Дегидратация метанола с получением диметилового эфира-исторически первый путь проведения данного синтеза. Этому процессу ещё с 1960-х годов было посвящено множество работ советских и ...
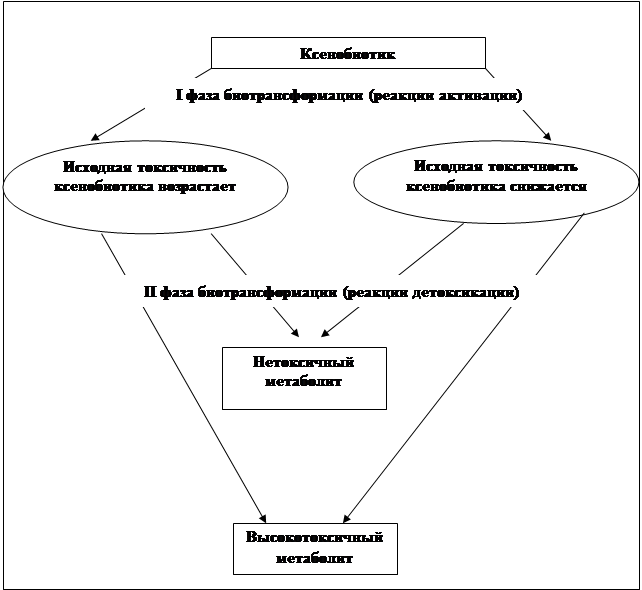
... затрат в рециркуляционных реакционно-ректификационных процессах с различной организацией подачи рецикла для реакции изомеризации типа АВ. Глава 2. Расчетно-аналитическая часть 2.1. Анализ стационарных состояний рециркуляционного реакционно-ректификационного процесса. В рециркуляционных схемах существуют различные варианты подачи рецикла. В данном случае рассматривается схема, ...
... процесс разделения нестабильных веществ можно проводить в холодильной камере. Выделенное соединение подвергают структурному химическому исследованию, а затем изучают его фармакологическое действие. Получение лекарственных веществ методом культуры тканей высших растений В нашей стране заготавливаются десятки тысяч тонн ЛРС. Однако потребность в БАВ, содержащихся в растениях, с каждым годом ...
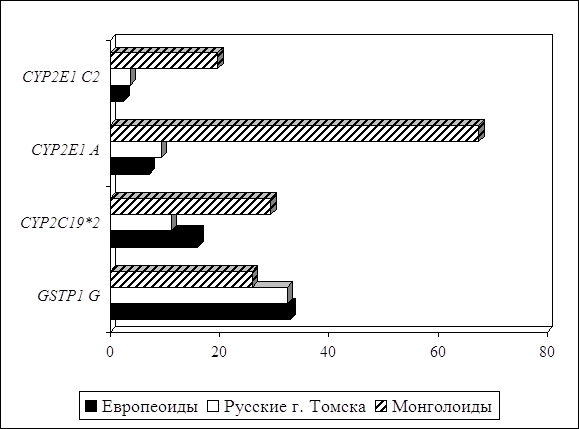
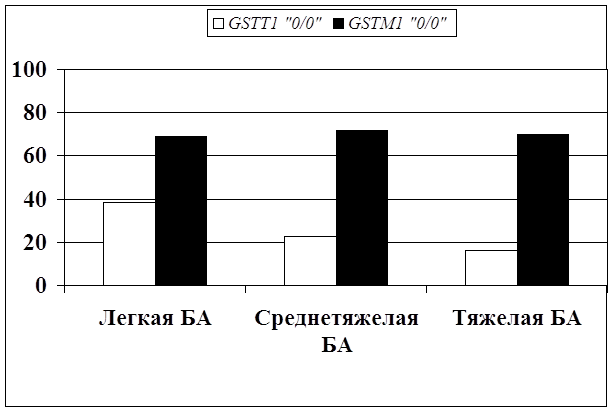
... препаратов. Установлена связь полиморфизма 313A>G гена GSTP1 с изменчивостью уровня аланинаминотрансферазы (р=0,021). 7. Выявлены различия в структуре генетической подверженности к бронхиальной астме и туберкулезу по генам ферментативной системы метаболизма ксенобиотиков: гены GSTM1, CYP2E1 и CYP2C19 связаны с бронхиальной астмой и значимыми для заболевания качественными и ...
0 комментариев