Навигация
7. Получение гидазепама
|
| Т.пл.=221oC |
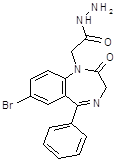
Стадия 1: Получение 7-бром-5- фенил-2,3-дигидро-1Н-1,4-бензодиазепин-2-он
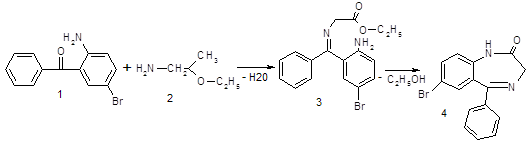
Стадия 2: Получение Na соли 7-бром-5- фенил-2,3-дигидро-1Н-1,4-бензодиазепин-2-он
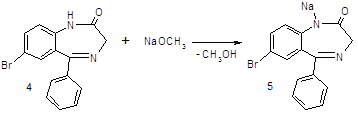
Стадия 3: Получение субстанции гидазепама
ОПИСАНИЕ ТЕХНОЛОГИИ
Из раствора 27 г. 5-бром-2-аминобензофенона (1) и 21г.хлоргидрата этилового эфира глицина (2) в 200мл. безводного пиридина при перемешивании в течении 12 ч. отгоняли 50 мл. пиридина. Одновременно добавляем к реакционной массе такое же количество пиридина. По окончании реакции пиридин отгоняют в вакууме при 40-50 oC. и к остатку прибавляют метиленхлорид и воду. Водный слой отделяют, подщелачивают и экстрагируют метиленхлоридом. Нерастворившуюся часть продукта отфильтровывают, промывают водой и сушат, а затем растворяют в кипящем бензоле. После охлаждения выпадают кристаллы вещества (4), которые присоединяем к основной массе продукта. Раствор вещества (4) в метиленхлориде промывают водой, сушат прокаленным сульфатом натрия и упаривают. Выход: (4) 14г. (65%),Т.пл.=221oC (из этилацетата)[4].
После получения вещества (4), мы при помощи метилата натрия переводим его в вещество (5), а затем при помощи гидразид хлоруксусной кислоты получаем конечный продукт (6)
Анализ ПРЕПАРАТОВ Методы анализа феназепама.Работа выполнена на опытных и серийных образцах феназепама, синтезированных на опытном заводе Физико-химического института АН УССР. Всего изучено более 30 серий препарата.
Реакции идентификации. Согласно требованиям ГФХ и ОСТов для идентификации препаратов подбираются реакции, при помощи которых можно доказать функциональные группы изучаемого соединения и отличить его от других лекарственных средств, близких по химическому строению. Для идентификации феназепама мы разработали следующие реакции:
1) Реакция на блокированную ароматическую аминогруппу. Феназепам гидролизовали при кипячении с разведенной соляной кислотой. На образовавшийся ароматический амин проводили реакцию диазотирования с нитритом натрия. Соль диазония сочетали с резорцином в щелочной среде и получали азокраситель красного цвета. При использовании b-нафтола вместо резорцина получали азокраситель оранжевого цвета.
Реакция образования азокрасителя используется для идентификации лекарственных средств бенздиазепинового ряда. Так, Британская фармакопея в 1973г. рекомендует ее для идентификации нитразепама и хлордиазепоксида, но для получения азокрасителя применяется другой реагент — гидрохлорид N-(1-нафтил)-этилендиамина.
2) Флуоресцентная реакция. С концентрированными кислотами (серной, хлорной и др.) феназепам образует соли зеленовато-желтого цвета, которые в ультрафиолетовом свете при длине волны 254 нм имеют яркую зеленую флуоресценцию. В связи с тем что феназепам практически не растворим в воде, реакцию с 57%-ным водным раствором хлорной кислоты мы проводили с раствором препарата в смеси хлороформа и 95%-ного спирта (1:1).
Структурные аналоги – (диазепам, нитразепам, медазепам) с указанными выше кислотами образуют соли, у которых в ультрафиолетовом свете флуоресценции иного цвета (зеленовато-голубая, голубая и голубовато-фиолетовая). Следовательно, реакцией с хлорной кислотой феназепам можно отличить от соединений, близких по химическому строению.
3) Специфическая реакция. При изучении физико-химических свойств феназепама мы обратили внимание на то, что при осторожном нагревании в сухой пробирке над пламенем горелки препарат плавится с образованием плава фиолетового или красно-фиолетового цвета.
Известно, что из многочисленного ассортимента лекарственных препаратов аналогичного цвета плавы образуются только при плавлении белого стрептоцида, сульгина и уросульфана. Поэтому было интересно изучить свойства плава феназепама и получить плавы соединений, близких по строению. Изучение показало, что плав феназепама имеет особенность: его окраска изменяется в зависимости от рН среды. Так, при добавлении раствора едкого натра красно-фиолетовая окраска раствора плава в 95%-ном спирте переходит в сине-фиолетовую, а при добавлении разведенной серной кислоты — в сине-зеленую, зеленую и желтую. По-видимому, при плавлении феназепама образуется соединение, имеющее свойства кислотно-основного индикатора.
У структурных аналогов феназепама (диазепама, нитразепама, медазепама) при плавлении образуются плавы преимущественно зеленого цвета и окраска спиртовых растворов этих плавов не изменяется в зависимости от рН среды.
Изложенное выше позволяет сделать вывод, что получение плава может быть использовано в качестве специфической реакции на феназепам, отличающей его от многих лекарственных средств, в том числе и от его структурных аналогов: диазепама, нитразепама и медазепама.
Методы определения примесей. Метод количественного определения. А. В. Богатский с сотрудниками изучил основные свойства бенздиазепинов и выявил, что группа С = N гетероциклической части молекулы сопряжена с кольцом бензола. Поэтому бенздиазепины являются слабыми основаниями, близкими по основности к пиридину и анилину. Феназепам как слабое основание не удается количественно определить ацидиметрическим методом в водной и спирто-водной средах. Нами разработана методика количественного определения феназепама путем неводного титрования 0,1-н. раствором хлорной кислоты в среде хлороформа и уксусного ангидрида (1:1). В качестве индикатора мы использовали кристаллический фиолетовый.
В уксусном ангидриде феназепам не растворяется, поэтому для его растворения использовали хлороформ. Кроме того, в присутствии хлороформа улучшается титрование. Как апротонный растворитель он уменьшает ионное произведение среды. Правильность перехода окраски индикатора в эквивалентной точке подтверждена потенциометрическим титрованием с применением хлорсеребряного и стеклянного электродов. Хлорсеребряный электрод заполняли 0,1-н. раствором перхлората лития в ледяной уксусной кислоте. Скачок потенциала совпадал с переходом окраски индикатора в желтый цвет.
Анализируемые образцы феназепама имели содержание не ниже 99%.
При помощи приведенных выше методов анализа мы установили, что феназепам устойчив при хранении в условиях комнатной температуры в защищенном от света месте в течение двух лет (срок наблюдения)[10].
По анализам проведенным с феназепамом, мы можем предположить что они подходят и к альпразоламу, гидазепаму, клобазаму и тофизопаму.
Анализ оксазепама. [12]Оксазепам содержит не меньше 98,5% и не больше 101,0% 7-хлор-3-гидрокси-5-фенил-2,3-дигидро-2Н-1,4-бензодиазепин-2-она, в пересчете на сухое вещество.
Кристаллический порошок белого или почти белого цвета. Очень мало растворимый в воде , растворим в 96% спирте и метиленхлориде.
Идентификация:
А. Растворы готовят в защищенном от яркого света месте и измеряют оптическое поглощение растворов сразу после приготовления.
10мг субстанции растворяют в 96% спирте и доводят объём раствора тем самым растворителем до 100,0мл. 10,0мл полученного раствора доводят 96% спиртом до объёма 50,0мл.(раствор А). 10мл раствора А доводят 96% спиртом до объёма 100,мл (раствор В). Ультрафиолетовый спектр поглощения полученного раствора А в области от 300нм до 350нм должен иметь два максимума по длине волн 316нм. Ультрафиолетовый спектр поглощения полученного раствора В в области от 220нм до 250нм должен иметь два максимума по длине волн 229нм. Удельный показатель поглощения в максимуме по длине волны 229нм должен быть приблизительно 1220 до 1300.
В. Инфрокрасный спектр поглощения субстанции, полученной в дисках должен соответствовать спектру ФСЗ оксазепема.
С. Хроматограммы полученные при испытании «Сопровождающие примеси» пересматривают в УФ-свете при длине волны 254нм. На хроматограмме испытуемого раствора (б) должно проявляться основное пятно на уровне пятна на хроматограмме раствора сравнения (а), соответственное ему по размеру.
Д. Около 20мг субстанции растворяют в смеси 5мл кислоты хлористоводородной и 10мл воды. Кипятят на протяжении 5 минут и охлождают. К полученному раствору добавляют 2мл раствора 1г/л натрия нитрита и выдерживают на протяжении 1 минуты. Добавляют 1мл раствора 5г/л сульфаминовой кислоты, перемешивают выдерживают на протяжении 1 минуты и добавляют 1мл раствора 1г/л нафтилендиамина дигидрохлорида, раствор окрашивается в красный цвет.
Испытание на чистоту:
Сопровождающиеся примеси.
Испытание проводят в защищенном от яркого света месте. Определение проводят методом тонкослойной хроматографии, используя как тонкий слой подхожий силикагель с флуоресцентным индикатором с оптимальной интенсивностью поглощения по длине волны 254нм. Перед использованием пластинку промывают метанолом. Когда фронт растворителя пройдёт 17см от линии старта, пластинку вытягивают из камеры и сушат на воздухе, потом при температуре от 100 до 105°С на протяжении 30 минут.
Испытываемый раствор. (а) 50 мг субстанции растворяют в ацетоне и доводят объём раствора тем же самым раствором до 10мл.
Испытываемый раствор. (б) 2 мл испытуемого раствора (а) доводят ацетоном до объёма 10мл.
Раствор сравнения. (а) 10мг ФСЗ оксазепама растворяют в ацетоне и доводят объём раствора тем же самым растворителем до 10,мл..
Раствор сравнения. (б) 10мг ФСЗ оксазепама и 10мг ФСЗ бромазепама растворяют в ацетоне и доводят объём раствора тем же самым растворителем до 10,мл..
Раствор сравнения.(с) 1мл раствора сравнения (б) доводят ацетоном до объёма 100мл.
Раствор сравнения.(д) 5мл раствора сравнения (с) доводят ацетоном до объёма 100мл.
На линию старта хроматографической пластинки наносят 100мкл (100мкг) испытуемого раствора (а), 20мкл (20мкг) испытуемого раствора (б), 20мкл (20мкг) раствора сравнения (а) и 20мкл (20мкг оксазепама и 20мкг бромазепама) раствора сравнения (б), 20мкл (0,2мкг) раствора сравнения (с), 20мкл (0,1мкг) раствора сравнения (д). Пластинку помещают в камеру с смесью растворителей объемов метанол – метиленхлорид. Когда фронт растворителей пройдёт 15см от линии старта, пластинку вытягивают из камеры, сушат на воздухе и пересматривают в УФ-свете по длине волны 254нм.
На хроматограме испытуемого раствора (а) любое пятно, кроме основного, не должно быть интенсивней пятна на хроматограмме раствора сравнения (с) (0,2%), и только одно пятно может быть интенсивней, чем пятно на хроматограмме раствора сравнения (д) (0,1%).
Результаты анализа являются вероятными, если на хроматограмме раствора сравнения (б) проявляются два чётко разделённых пятна.
Потеря массы при высушивании. Не более 0,5% 1,000г субстанции сушат при температуре от 100°С до 125°С при остаточном давлении не больше 0,7кПа.
Сульфатная зола. Не больше 0,1%. Определение проводят с 1,0г субстанции.
Количественное определение:
250г субстанции растворяют в смеси 10мл ледяной уксусной кислоты и 90мл уксусного ангидрида и титруют 0,1М раствором кислоты хлорной потенциометрически.
1мл 0,1М раствора хлорной кислоты соответствует 28,67мг С15Н11ClN2O2
Хранение. В плотно укупоренном контейнере, в защищенном от света месте.
Анализ мепротана и его лекарственных форм. [13]Описание: белый кристаллический порошок без запаха или со слабым запахом.
Растворимость: мало растворим в воде и эфире, легко растворим в 95% спирте и ацетоне.
Подлинность: 1. 0,1г препарата помещают в пробирку, прибавляют 3мл раствора едкого натрия и кипятят. Выделяется аммиак, обнаруживаемый по запаху и по посинению влажной красной лакмусовой бумаги.
0 комментариев