Навигация
Прогрессирующие мышечные дистрофии
150673
знака
3
таблицы
7
изображений
ГОУ ВПО Волгоградский государственный медицинский университет Росздрава
Кафедра детских болезней педиатрического факультета
Прогрессирующие мышечные дистрофии
Выполнено:
ст. 502 группы МБФ
Служенко М.О.
Проверено:
доцент Марушкин Д.В.
Волгоград, 2010
Содержание
Введение
1. Эпидемиология
2. Этиология
3. Патогенез
4. Клиническая классификация
5. Клиника
6. Диагностика
7. Лечение
8. Профилактика
Список литературы
Введение
Прогрессирующие мышечные дистрофии – это группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями [Гусев Е.И., Никифоров А.С., 2007].
1. Эпидемиология прогрессирующих мышечных дистрофий
Первое сообщение о прогрессирующей мышечной дистрофии было опубликовано в России в 1895 г. врачом В.К. Ротом, который назвал заболевание мышечной сухоткой. Заболевание описано во всех странах мира. Частота 3,3 на 100 000 населения, 14 на 100 000 родившихся. В подавляющем большинстве случаев болеют мальчики. Случаи заболевания у девочек крайне редки, хотя и возможны при кариотипе ХО и при структурных аномалиях хромосом (Хр21.2, ген DMD дистрофина), в 35—40% случаев носит семейный характер [Гринио Л.П., Агафонов Б.В. 1997].
Частота прогрессирующей мышечной дистрофии Дюшенна варьирует от 9,7 до 32,6 на 100 000 живорожденных мальчиков. Высокая распространенность заболевания в популяции в значительной мере связана с высокой частотой новых мутаций [Bejaoui K., Hirabayashi K., Hentati F. et al., 1995].
Офтальмоплегическая мышечная дистрофия относится к числу редких заболеваний, частота встречаемости в Европе составляет 1:100 000—200 000 человек [Гусев Е.И., Коновалов А.Н., Скворцова В.И., Гехт А.Б., 2009]. Однако в некоторых этнических группах и территориальных группах с «эффектом основателя» частота ОФМД намного выше, например во франко-канадской популяции — 1:1000 человек, у евреев Бухары, — 1:600. Также описаны выборки больных с офтальмоплегической мышечной дистрофией в более чем 30 странах на всех пяти континентах [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Популяционная частота прогрессирующей мышечной дистрофии Эрба - Рота составляет 1,2-2,5 случая на 100 000 населения [Гехт Б.М. и Ильина Н.А., 1998].
Частота встречаемости плече-лопаточно-лицевой миодистрофии Ландузи — Дежерина составляет 2,9 на 100 000 населения [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].
2. Этиология
Причиной являются генетически обусловленные дефекты метаболизма или структуры мышечной ткани, приводящие к атрофии мышц, разрастанию соединительной ткани и увеличению жировой клетчатки (псевдогипертрорфии) [Гусев Е.И., Никифоров А.С., 2007].
Табл. 1 Гены, ответственные за возникновение прогрессирующих мышечных дистрофий [Nevo Y., Muntoni F., Sewery C. et al., 1998]
| Название | Английская аббревиатура, синонимы | Тип наследования | Локализация гена | Ген | Белковый продукт гена |
| ПМД Дюшенна | DMD | ХР | Xp21.2 | DMD (DYS) | Дистрофин |
| ПМД Беккера | BMD | ХР | Xp21.2 | DMD (DYS) | Дистрофин |
| ПМД Эмери-Дрейфуса со сцепленным с полом наследованием | EDMD, скапуло - перонеальная форма, плече - лопаточно - перонеальная | ХР | Xq28 | Ген Эмерина, EDMD | Эмерин |
| ПМД Эмери-Дрейфуса, аутосомно-доминантный тип | EDMD2, скапуло - перонеальная форма, плече - лопаточно - перонеальная | АД | 1q21.2 | Ген Ламина А/C (LMNA/C) | Ламин А/С |
| ПМД Эмери-Дрейфуса, аутосомно-рецессивный тип | EDMD2, скапуло - перонеальная форма, плече - лопаточно - перонеальная | АР | 1q21.2 | Ген Ламина А/C (LMNA/C) | Ламин А/С |
| ПМД Ландузи-Дежерина | FSHD1, лице - плече - лопаточная форма | АД | 4q35 | FSHMD1A | - |
| Группа конечностно-поясных ПМД | |||||
| Окулофарингеальная форма, аутосомно-доминантный тип | OPMD | АД | 14q11.2-13 | PABP2 | Полиаденилин - ассоциированный белок |
| Окулофарингеальная форма, аутосомно-рецессивный тип | OPMD | АР | 14q11.2-13 | PABP2 | Полиаденилин - ассоциированный белок |
Различные формы прогрессирующих мышечных дистрофий могут наследоваться аутосомно-доминантно, аутосомно-рецессивно, рецессивно, сцепленно с Х-хромосомой. Различные формы прогрессирующих мышечных дистрофий отличаются разным типом наследования, вариабельностью возраста начала заболевания, преимущественной локализацией поражения мышц и другими признаками [Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др., 1999].
Миодистрофии Дюшенна и Беккера являются аллельными вариантами экспрессии единого генетического дефекта в локусе Р21 Х-хромосомы. Ген является самым большим из известных на сегодняшний день и имеет очень сложную молекулярную организацию; состоит из 79 экзонов (информативно значимых участков ДНК). В 60—65 % случаев мутация представляет собой делецию гена дистрофина, а в 5—10 % — его дупликацию. Встречаются и точковые мутации гена (до 30 % случаев) [Самуэльс М., 1997]. Высокая частота спорадических случаев миодистрофий Дюшенна и Беккера обусловлена чрезвычайно высокой частотой спонтанных мутаций гена, возможно, отчасти из-за его "гигантского" размера [Свердлов Е.Д., 1997]. С локусом Р21 Х-хромосомы ассоциированы также другие, редко встречающиеся, клинические фенотипы: семейная Х-сцепленная миалгия с крампи, синдром Мак-Леода (повышение уровня КФК, акантоцитоз), квадрицепс-миопатия. Последняя является наиболее мягкой формой и характеризуется медленным профессированием слабости четырехглавых мышц бедра, гипертрофией голеней и повышением КФК. При миодистрофий Дюшенна уровень дистрофина не превышает 3 % от нормального, тогда как при болезни Беккера он колеблется от 3 до 20 % [Гехт Б.М. и Ильина Н.А., 1998].
Этиология псевдогипертрофической формы Дюшенна.
Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна, Xp21.2, ген DMD дистрофина) — возникает в результате дефектов гена, кодирующего белок дистрофин. Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов. Мутации в гене дистрофина вызывают клинически явно различающиеся формы миодистрофий [Евтушенко С.К., Садеков И.А. 1994].
Некоторые авторы предполагают, что различия в клинике дистрофинопатий могут определяться характером мутаций, из которых одни приводят к сдвигу рамки считывания (в результате чего синтез соответствующего белка практически невозможен), тогда как другие повреждают ген, но не нарушают рамку считывания (результатом чего становится синтез измененного белка, частично способного к функционированию) [Евтушенко С.К., Садеков И.А. 1994]. Первый вид мутаций обычно связывают с тяжелым течением болезни, второй - с более мягким. Данное предположение, известное как гипотеза Монако, нашло подтверждение в ряде публикаций [Гринио Л.П., Агафонов Б.В., 1997].
Вместе с тем накопление сведений о мутациях в гене дистрофина и изучение клинико-генетических корреляций при дистрофинопатиях способствовали выявлению случаев, трудно объяснимых с позиций гипотезы Монако или других гипотез, постулирующих жесткую связь особенностей клиники с глубиной повреждения функции белка. Еще в 1988 г. было опубликовано сообщение о нескольких больных, у которых выявлялись делеции экзонов 3-7 гена дистрофина и соответственно сдвиг рамки считывания, но при этом заболевание протекало в мягкой форме - как миодистрофии Беккера [Шишкин С.С., 1997]. Даже если бы у пациентов только отсутствовал данный участок последовательности дистрофина (не говоря уже о нарушении рамки считывания для остальной части гена), можно было бы ожидать серьезного нарушения функции белка, так как, по имеющимся сведениям, именно эта часть молекулы дистрофина обеспечивает его связывание с актином [McKusik V., Amberger J., 2003]. Однако болезнь в ряде случаев протекала относительно доброкачественно. Более того, известен пациент с делецией экзонов 3-9 в дистрофиновом гене, который до 60-летнего возраста и не подозревал о своей болезни, а в 67 лет сохранял способность к самостоятельной ходьбе [Ahn A.H., Kunkel L.M., 1998]. Авторы, описавшие этот уникальный случай, предположили, что, следовательно, возможны делеции в функционально важной области дистрофинового гена, которые обеспечивают состояние с длительным бессимптомным течением и без существенного сокращения продолжительности жизни [Kaplan J.C., Fontaine В., 1999].
В других публикациях отмечалось, что крупные делеции, захватывающие 26% (экзоны 21-44) и даже 40% последовательности дистрофинового гена, иногда обусловливают позднее начало и очень мягкое течение болезни - такие пациенты в возрасте 55 и 60 лет сохраняли определенную двигательную активность [Шаховская Н.И., 2000].
Этиология офтальмоплегической мышечной дистрофии Кило-Невина.
В 1998 г. изолировали на хромосоме 14q11.2-13 ген поли-(А)-связывающего белка 2 (PAPB2, PABPN1), ответственный за синтез ядерного белка PABP2, служащего фактором полиаденилирования мРНК, и идентифицировали мутацию, заключающуюся в увеличении числа копий тринуклеотидных GCG-повторов в 1-м экзоне гена. В норме ген содержит шесть тандемных копий повторов GCG, а у больных их число достигает до 8—13. В некоторых популяциях экспансия числа тринуклеотидных повторов происходит за счет простого добавления GCG-повторов, в других — вместе с экспансией GCG-повторов происходит GCA-вставка.
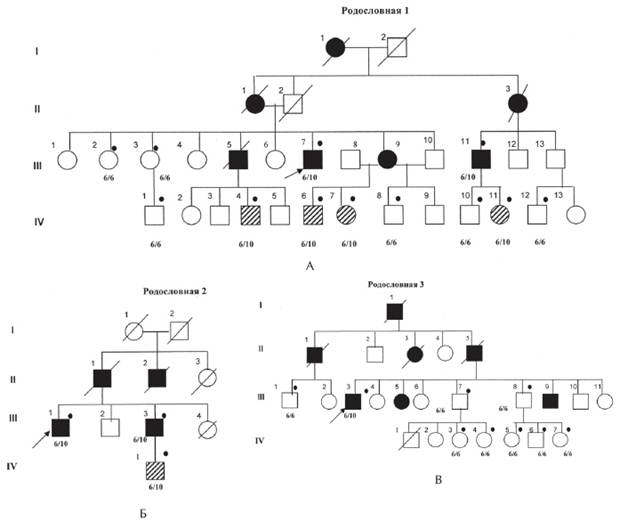
Рисунок 1. Рис. 1. А, Б, В. Родословные семей с офтальмоплегической мышечной дистрофии Кило-Невина с результатами ДНК-диагностики [Codere F., Brais B., Rouleau G., Lafontaine E., 2001].
Механизм развития экспансии тандемных повторов при офтальмоплегической мышечной дистрофии до сих пор неясен; высказываются предположения, что он может быть связан с неравным кроссинговером, разновидностью гомологичной рекомбинации, происходящей в зародышевых клетках во время мейоза или иногда митоза.
Также описаны случаи точечной мутации в гене PABPN1 22 и случаи гомозиготного носительства «промежуточного» аллеля гена, имеющего 7 GCG-повторов с аутосомно-рецессивным типом наследования с развитием более тяжелой клинической картины и более ранним дебютом симптомов заболевания [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Примечание. 6/6; 6/10 — результаты ДНК-анализа на мутацию в гене PABPN1; Закрашенный кружок —обследованные пациенты; закрашенный квадрат — больной с ОФМД, пустой квадрат — клинически здоровый; заштрихованный квадрат — клинически здоровый носитель мутации в гене PABPN1. I, II поколение — умершие родители, III поколение — больные и их сибсы, IV — дети, не достигшие возраста начала заболевания
Этиология конечно-поясной формы Эрба-Рота.
Известно не менее 9-ти локусов, ответственных за прогрессирующую мышечную дистрофию Эрба-Рота. Чаще всего вовлечен локус 15q15-q21.1 [McKusik V., Amberger J., 2003], реже вовлекается один из локусов, расположенных в коротком плече хромосомы 2 [Bashir R. et al., 1994], еще реже заболевание связывают с локусом 13q [Lim L.E., Duclos P., Broux O. et al., 1995].
В международной литературы конечностно-поясные формы обозначаются аббревиатурой LGMD (limb-girdle muscular dystrophy) c указанием типа, например LGMD 1A. Арабской цифрой 1 обозначаются типы с аутосомно-доминантным типом наследования, 2 – аутосомно-рецессивные формы [Шишкин С.С., Н.И. Шаховская, И.Н. Крахмалева, 2002]. Как видно из представленной таблицы 2, конечностно-поясные формы прогрессирующих мышечных дистрофий – это целая группа генетически гетерогенных заболеваний, объединенных общей клинической картиной: прогрессирующая проксимальная мышечная слабость и гипотрофии, симптомы «крыловидных лопаток», «утиной походки», поясничный гиперлордоз. LGMD2A соответствует ювенильной конечностно-поясной форме прогрессирующих мышечных дистрофий Эрба-Рота [Умаханова Р. С. С. Жилина Г. Р. Мутовин, 2005].
Табл.2 Гены, дефекты которых ответственны за возникновение миодистрофии Эрба-Рота (пояснично-конечностная миодистрофия или LGMD) [Умаханова Р. С. С. Жилина Г. Р. Мутовин, 2005].
| Типы | Особенности | Тип наследования | Локализация гена | Ген | Белковый продукт гена |
| 1A | АД | 5q31 | Миотилин | ||
| 1B | Аллельная форма ПМД Эмери-Дрейфуса | АД | 1q21 | Ламин А/С | |
| 1C | АД | 3p25 | CAV3 | Кавеолин-3 | |
| 1D | АД | 7q | |||
| 1E | Дилатационная кардиомиопатия | АД | 6q23 | ||
| 1F | АД | 7q32 | |||
| 1G | АД | 4p21 | |||
| 2A (Эрба) | Начало 2-45 лет, в среднем 14-20 лет. | АР | 15q15.1-q15.3 | CAPN3 | Кальпаин-3 |
| 2B | Аллельная форма – дистальная дистрофия Миоши | АР | 2p13.1 | Дисферлин | |
| 2C | АР | 13q12 | SGCG |
| |
| 2D | АР | 17q21 | SGCA |
| |
| 2E | АР | 4q12 | SGCB |
| |
| 2F | АР | 5q33 | SGCD |
| |
| 2G | АР | 17q11-12 | Телетонин | ||
| 2H | АР | 9q31-33 | TRIM32 | ||
| 2I | Аллельная форма мерозиновой миопатии (ламинин-2) и конгенитальной мышечной дистрофией с мышечными гипертрофиями и нормальной ЦНС | АР | 19q13.3 | FKRP | Фукутин-связанный белок |
| 2J | Аллельная форма дилатационной кардиомиопатии 1G и Finnish дистальной миопатии | АР | 2q31 | Титин | |
| 2K | С умственной отсталостью | АР | 9q34 | POMT1 |
Экспрессивность генов конечно-поясной формы Эрба-Рота значительно варьирует не только в популяции, но даже в пределах одной пораженной семьи, что, по-видимому, и определяет различную тяжесть и прогрессирование миодистрофического процесса у больных, а также существование относительно доброкачественных или злокачественных форм патологии [Бадалян Л.О., 2008; Вельтищев Ю.Е. и соавт., 1998].
Этиология миодистрофии Дрейфуса-Хогана
В 1990-х годах последовательно были идентифицированы ген эмерина в локусе Хq28, ответственный за Х-сцепленную миодистрофию Дрейфуса-Хогана, и ген ламинов LMNA в локусе 1q21.2, вызывающий аутосомно-доминантную форму, вслед за чем появились многочисленные верифицированные наблюдения обоих генетических вариантов [Бадалян Л. О., Темин П.А., Калинин В.А. и др., 1990; Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др., 2004].
Клиническая общность Х-сцепленной и аутосомно-доминантной форм неслучайна: она обусловлена тесным функциональным взаимодействием ламинов А/С и эмерина - мембранных белков, участвующих в образовании каркаса ядерной оболочки. При общем основном симптомокомплексе клиническая картина обеих генетических форм миодистрофии Дрейфуса-Хогана, особенно аутосомно-доминантной, демонстрирует разнообразие по возрасту начала, темпам прогрессирования, выраженности отдельных симптомов и тяжести болезни в целом, активности КФК, наличию атипичных признаков. Меж- и внутрисемейное клиническое разнообразие отмечалось уже в первых описаниях Х-сцепленной и аутосомно-доминантной миодистрофии Дрейфуса-Хогана вариантов [Бадалян Л. О., Темин П.А., Калинин В.А. и др., 1990].
В международных базах данных зарегистрировано более 100 мутаций гена эмерина и около 200 мутаций LMNA (большинство - при миодистрофии Дрейфуса-Хогана), действительное число мутаций несомненно больше, поскольку не все исследователи регистрируют свои находки в базах данных. Преобладают миссенс-мутации. Преобладающих по частоте («мажорных») мутаций нет, большинство мутаций обоих генов встречаются в единичных семьях, но некоторые описаны неоднократно. К ним относится мутация Arg249Gln в экзоне 4 гена LMNA, выявленная у больных с разными фенотипами. Мутация Arg249Gln возникает de novo, что позволяет предполагать наличие мутационной «горячей точки» в гене LMNA [Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др., 2004]. Так же имеет место мутация Arg377His в экзоне 6 [Мальмберг С.А., Петрухин А. С., Широкова В.И., 2000].
Нормальный биохимический продукт гена эмерин – представляет собой обогащенный аминокислотой (серином) белок, состоящий из 254 аминокислот. Эмерин экспрессируется преимущественно в скелетных, гладких мышцах и кардиомиоцитах; ему принадлежит значительная роль в организации клеточного цитоскелета и везикулярного транспорта. В сердечной мышце эмерин обеспечивает межклеточную адгезию и осуществление контактов между кардиомиоцитами. Типичная мутация представлена делецией гена и приводит к прекращению синтеза эмерина [Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др., 1999].
Этиология миодистрофии Бетлема
Редкая доброкачественная миодистрофия, наследуемая по аутосомно-доминантному типу. Установлена генетическая гетерогенность болезни: один из генов картирован в локусе 21q22, другой — 2q37. В результате мутаций нарушается синтез субъединиц коллагена VI типа, который обеспечивает связь базальной мембраны с гликопротеинами внеклеточного матрикса [Вельтищев Ю.Е., Темин П.А., 1998].
Похожие работы
... Hungary. – CD. – A0034. Автор провів аналіз клініко-ортопедичних даних у хворих на ПМД, який було покладено в основу написання тезів. АНОТАЦІЯ Зима А.М. Діагностика та ортопедичне лікування різних форм прогресуючої м’язової дистрофії. Рукопис. Дисертація на здобуття наукового ступеня кандидата медичних наук за спеціальністю 14.01.21 – травматологія та ортопедія ДУ «Інститут травматології та ...
... ревматизма обусловила значительное снижение заболеваемости — до 0Д8 на 1000 детского населения. В разработку проблемы детского ревматизма внесли большой вклад отечественные педиатры В. И. Молчанов, А. А. Кисель, М. А, Скворцов, А. Б. Воловик, В. П. Бисярина, А. В. Долгополова и др. Эпидемиология, Установлена связь между началом заболевания и перенесенной стрептококковой инфекцией, в основном в ...
... 036.При инфаркте в бассейне передней артерии сосудистого сплетения (передняя ворсинчатая) не бывает #а)гемиплегии #б)гемианестезии *#в)афазии #г)вазомоторных нарушений в области парализованных конечностей #д)гемианопсии 037.Препараты наперстянки и строфанта при декомпенсации дисциркуляторной энцефалопатии назначают #а)для нормализации сердечного ритма ...
... КМГ, которая в начале может быть парциальной. Размеры сердца и степень увеличения отдельных камер в большой сетпени зависят от характера порока. См. Методическое пособие "Дифференциальный диагноз при шумах сердца". Синдром Марфана. Комплекс наследственных аномалий (наследование аутосомно-доминантное), связанных с поражением соединительной ткани. Типичны изменения скелета, включащие ненормально ...
0 комментариев