Навигация
5. Клиника
Первые признаки болезни проявляются нарастающей слабостью тех или иных групп мышц, утомляемостью при легких физических нагрузках, симметричными атрофиями мышц [Евтушенко С.К., Садеков И.А. 1994].
Характерными симптомами прогрессирующих мышечных дистрофий являются мышечная слабость и атрофия мышц, которые могут проявляться в различные возрастные периоды, но чаще развиваются в детском и юношеском возрасте [Гаусманова - Петрусевич И., 2001]. Дети поздно начинают ходить, быстро утомляются, неуклюжи в ходьбе, спотыкаются при беге, часто падают, с трудом поднимаются по лестнице. Двигательные нарушения постепенно прогрессируют. Возникает миопатическая утиная походка. В случае поражения мышц тазового пояса и конечностей затруднен переход из горизонтального положения в вертикальное; при поражении дистальных групп мышц ног появляется петушиная походка. Стойкость и нарастание двигательных нарушений позволяют диагностировать миодистрофию уже на ранних стадиях заболеваниях. При обследовании больного обнаруживают генерализованную или локальную атрофию мышц. Локальная атрофия мышц выявляется лишь на ранних стадиях заболевания, по мере прогрессирования патологического процесса атрофия мышц приобретает генерализованный характер вплоть до мышечной кахексии. Атрофированные мышцы истончены, дряблые при пальпации, однако следует отметить, что наряду с атрофией мышц выявляется псевдогипертрофия (замещение атрофированных мышц жировой клетчаткой, соединительной тканью). Миодистрофический процесс сопровождается поражением соединительной ткани, миосклерозом, развитием сухожильно-связочных ретракций, ограничением объема движений в суставах, укорочением пяточного (ахиллова) сухожилия, контрактурами. Одновременно с развитием мышечных атрофий снижаются сухожильные рефлексы, в первую очередь коленные [Вельтищев Ю.Е., Темин П.А., 1998].
Поражение мышц плечевого пояса приводит к ограничению движений в плечевых суставах. Больные не могут поднять руки выше горизонтального уровня, в то время как объем движений в локтевых и лучезапястных суставах и сила мышц длительное время остаются сохранными. При попытке поднять больного подмышки его голова как бы проваливается в плечи — симптом «свободных надплечий». Лопатки отстают от туловища — симптом «крыловидных лопаток». При поражении мышц тазового пояса возникают затруднения при подъеме на лестницу, вставании из положения сидя. При этом больной оказывает себе помощь, опираясь на посторонние предметы, встает в несколько этапов («лесенкой»). Изменяется походка: она становится переваливающейся, раскачивающейся - «утиная» походка. Атрофия косых мышц живота приводит к развитию «осиной» талии. Слабость длинных мышц спины нарушает осанку, приводит к искривлению позвоночника и выпячиванию живота [Гаусманова - Петрусевич И., 2001].
Поражение мышц костей и стоп сопровождается их слабостью. Походка больных становится своеобразной. Для того чтобы не зацепиться носком отвисающей стопы за пол, больные вынуждены высоко поднимать голень— «петушиная» походка [Евтушенко С.К., Садеков И.А., 1994].
При слабости и атрофии мышц лица отмечается отсутствие морщин на лбу (симптом «полированного лба»). Наблюдается гипомимия: больные не могут плотно зажмурить глаза, надуть щеки, вытянуть губы в трубочку и т. д. В некоторых случаях вследствие замещения губных мышц соединительной и жировой тканью губы утолщаются (напоминают губы тапира) [Hoffmann E.P., Kunkel L.M., Angelini C. et al., 2009].
При поражении наружных глазных мышц отмечается ограничение объема движения глазных яблок; иногда они становятся полностью неподвижными [McNally E., Passos-Bueno R., Bonnemann C.G. et al., 1996].
Если в патологический процесс вовлекаются мышцы глотки и гортани, возникает осиплость голоса и нарушается акт глотания. Поражение межреберных мышц ведет к дыхательной недостаточности и заболеваниям легких и сердца [Minetti C., Sotgia F., Bruno C. et al., 1998].
При неврологическом обследовании больных с прогрессирующими мышечными дистрофиями наряду с ограничением объема движений, снижением силы мышц и их атрофией выявляются мышечная гипотония, снижение или полное отсутствие сухожильных рефлексов [Moreira E., Vainzof M., Marie S. et al., 1997].
Темп прогрессирования патологического процесса зависит от формы заболевания и индивидуальных особенностей организма. В стадии выраженных нарушений вследствие атрофии мышц и отсутствия движений могут формироваться контрактуры (тугоподвижность или невозможность движения в суставах) [Minetti C., Sotgia F., Bruno C. et al., 1998].
Большинство форм прогрессирующих мышечных дистрофий не сопровождается снижением интеллекта. Больные критически относятся к своему дефекту. Иногда наблюдаются выраженные эмоциональные нарушения в виде повышенной раздражительности, подавленности настроения, замкнутости. Большинство больных успешно обучается по программе массовой школы [Muntoni F., Mateddu A., Marchei F. et al., 1993].
Исключение составляют больные псевдогипертрофической формой. При этой форме наблюдается выраженное снижение интеллекта. Данный вариант прогрессирующей мышечной дистрофии наследуется рецессивно, сцеплен-но с У-хромосомой. Основную массу больных составляют мальчики. Наряду с прогрессирующими атрофиями и слабостью мышц плечевого и тазового пояса у больных наблюдаются псевдогипертрофии (разрастания соединительной ткани, особенно в области икроножных мышц) и эндокринные нарушения (чаще ожирение). Некоторая задержка развития психических функций отмечается уже в первые годы жизни. Дети малоэмоциональны. Речь развивается с запозданием и носит примитивный характер. Отсутствует абстрактное мышление. Навыки опрятности и самообслуживания формируются с трудом. Интеллект обычно классифицируется как тяжелая дебильность или имбецильность; реже наблюдается идиотия [Гаусманова - Петрусевич И., 2001].
Псевдогипертрофическая злокачественная миодистрофия Дюшенна
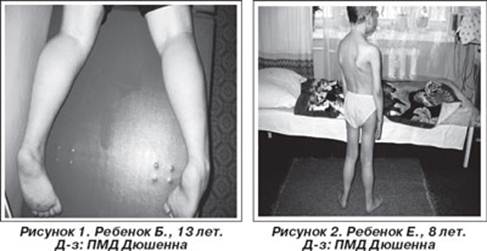
Первое наблюдение псевдогипертрофической миопатии принадлежит E.Meryon (1852), опубликовавшему в статье "К вопросу о жировой и гранулярной дегенерации мышц" семейный случай болезни у 4 братьев с аномальным увеличением икроножных мышц и контрактурами конечностей. G.Duchenne в 1861 г. описал больного с "псевдогипертрофическим мышечным параличом", обратив внимание на необычное сочетание увеличения икроножных мышц с прогрессирующей мышечной слабостью. В 1868 г. G.Duchenne опубликовал серию статей в журнале "Архив общей медицины", где представил систематизированный анализ болезни [Moreira E., Vainzof M., Marie S. et al., 1997].
Проявляется в возрасте 2—5 лет. Течение быстро прогрессирующее, злокачественное. Обездвиженность больных, как правило, наступает в возрасте 14—15 лет, смерть наступает в возрасте 15—18 лет, больные редко живут более 25 лет. К 8-10 годам большинство детей нуждается в ортопедических аппаратах; к 12 годам большинство детей не могут ходить. Первые признаки заболевания проявляются в 1-3 года жизни слабостью мышц тазового пояса. Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают. В 2-3 года появляются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке - длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, с положения на корточках или со стула. Вставание происходит поэтапно с активным использованием рук - «взбирание лесенкой» или «взбирание по самому себе». Типичные жалобы родителей — это ходьба детей на пальцах и частые падения. Задержка темпов двигательного развития часто обнаруживается ретроспективно при анализе анамнестических сведений. Ранние симптомы подкрадываются незаметно. Недостаточную, по сравнению со сверстниками, подвижность ребенка, его двигательную пассивность часто относят к особенностям темперамента и характера [Гринио Л.П,. 1998].
Псевдогипертрофия икроножных мышц создает обманчивое впечатление о сохранности мышечной силы и даже радует родителей. Псевдогипертрофии мышц могут развиваться также в ягодичных, дельтовидных мышцах, мышцах живота и языка. Дети могут не привлекать внимания специалиста до тех пор, пока проксимальная мышечная слабость не станет настолько выраженной, что затруднит вставание ребенка с пола и определит утиный тип ходьбы и использование миопатических приемов "взбирания по себе" (симптом Говерса). Ретракция пяточных (ахилловых) сухожилий не позволяет больному полноценно опираться на пятки, что определяет ходьбу на пальцах. На протяжении детства двигательная сила постепенно снижается. Двигательные функции выглядят относительно стабильными между 3 и 6 годами жизни. В большинстве случаев возможность ходьбы и подъема по лестнице сохраняется до 8-летнего возраста. Между 3 и 8 годами происходит нарастающее укорочение пяточных сухожилий и формируются сгибательные контрактуры в голеностопных суставах, развиваются поясничный гиперлордоз, кифосколиоз грудного отдела позвоночника. Нарастают атрофии мышц бедра, тазового пояса, а затем плечевого пояса, спины и проксимальных отделов рук. Атрофии мышц всегда симметричны. Нередко атрофии мышц маскируются хорошо развитой подкожной жировой клетчаткой. Изменения костной системы не ограничиваются лишь сколиозом: часто развиваются деформации грудной клетки и стоп, диффузный остеопороз. Ухудшение походки ведет к тому, что дети все чаще падают. Вначале атрофии локализуются в проксимальных группах мышц нижних конечностей - мышцах тазового пояса, бедер, а через 1-3 года быстро распространяются в восходящем направлении на проксимальные группы мышц верхних конечностей - плечевой пояс, мышцы спины. Вследствие атрофии появляются лордоз, «крыловидные» лопатки, «осиная» талия. Типичным, «классическим» симптомом заболевания является псевдогипертрофия икроножных мышц. При пальпации мышцы плотные, безболезненны. У многих больных в результате селективного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и сухожильные ретракции. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Глубокие рефлексы изменяются с различной последовательностью. В ранних стадиях болезни исчезают коленные рефлексы, позже - рефлексы с двуглавой и трехглавой мышц. Ахилловы рефлексы длительное время остаются сохранными. Характерны симметричная и неуклонно прогрессирующая слабость в мышцах бедер и плечевого пояса, затрудняющая движения при подъеме, беге, прыжках, поясно-конечностная атрофия мышц, преимущественно мышц тазового пояса и бедер, истинная гипертрофия или псевдогипертрофия икроножных мышц, ранние сухожильно-связочные ретракции (укорочение сухожилий и связок), контрактуры крупных суставов. Коленные рефлексы рано исчезают, ахилловы рефлексы сохраняются [Muntoni F., Mateddu A., Marchei F. et al., 1993].
Одной из отличительных особенностей миодистрофии Дюшенна является сочетание данной формы с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем). Костно-суставные нарушения характеризуются деформациями позвоночника, стоп, грудины. На рентгенограммах обнаруживают сужение костномозгового канала, истончение коркового слоя длинных диафизов трубчатых костей [Novakovic I., Todorovic S., Apostolski S. et al., 1998].
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На ЭКГ регистрируются изменения миокарда (блокада ножек пучка Гиса и др.). Нейроэндокринные нарушения встречаются почти у половины пациентов. Чаще других даются синдром Иценко-Кушинга, адипозогенитальная дистрофия Бабинского-Фрелиха [Страхова О.С., Белозерова Ю.М., Темин П.А., 1999].
Установлено, что при мышечной дистрофии Дюшенна сердечно-сосудистая система вовлекается в патологический процесс достаточно часто и рано. Около 73% больных с данной нозологией имеют различные проявления кардиальной патологии. Причиной сердечно– сосудистой патологии является генетически детерминированный недостаток дистрофина в кардиомиоцитах [Страхова О.С., Белозерова Ю.М., Темин П.А., 1999].
Отсутствие четкой корреляции между тяжестью поражения скелетных мышц и наличием выраженной кардиомиопатии у пациентов с прогрессирующей мышечной дистрофии Дюшенна предопределило необходимость обратить особое внимание на исследование маркеров вовлечения сердечной мышцы в патологический процесс. Оказалось, что делеции гена дистрофина являются не единственной причиной поражения мышечной ткани у пациентов с прогрессирующей мышечной дистрофии Дюшенна. В настоящее время ученые выделяют три основных причины: дефицит дистрофина, обусловленный генетическим дефектом; дефицит дистрофин- ассоциированного гликопротеина (молекулярная масса 50 кДа) или других дистрофинассоциированных белков, наличие особого генетического варианта строения ангиотензин- конвертирующего фермента. Сердечная мышца может поражаться как вследствие всех трех причин, так и их комбинаций. Например, дефицит дистрофинассоциированного гликопротеина у пациентов с прогрессирующей мышечной дистрофией Дюшенна может наблюдаться исключительно в кардиомиоцитах, в то время как в склетной мышечной ткани его содержание будет нормальным . Обнаружение дефицита дистрофинассоциированных белков при исследовании биоптата сердечной мышцы является предиктором развития тяжелой кардиомиопатии. Особое внимание последние годы уделяется строению ангиотензин-конвертирующего фермента. По мнению Kasper EK с соавт. тяжесть кардиомиопатии при прогрессирующей мышечной дистрофии Дюшенна взаимосвязана особенностями строения ангиотензин- конвертирующего фермента у больного. Выявление маркеров вовлечения сердечной мышцы в патологический процесс позволяет ответить на исключительно важный практический вопрос – почему кардиомиопатия может наблюдаться у пациентов с легкими вариантами поражения скелетных мышц, а также возможность дебюта заболевания с кардиомиопатии [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
По данным А.Oldfors, начальные проявления кардиальной патологии у больных возникают уже в раннем возрасте и прогрессируют с годами. В отдельных случаях, у детей 3-5 лет, в клинической картине заболевания могут преобладать кардиальные симптомы, а симптомы мышечной дистрофии могут быть маскированными [Novakovic I., Todorovic S., Apostolski S. et al., 1998].
Низкая физическая активность пациентов с прогрессирующей мышечной дистрофии Дюшенна, относительно быстрая утрата способности к самостоятельной ходьбе, снижающая нагрузку на миокард, а также недостаточная нацеленность родителей на выявление кардиальных жалоб (основное внимание обращается прежде всего на двигательные нарушения), приводят к тому, что менее 15% детей до 14 лет, имеющих поражение мышцы сердца, активно обращаются к кардиологу. В то время как по данным целевых исследований у детей, не предъявляющих кардиальных жалоб, поражение мышцы сердца выявляется у 25% в возрасте до 6 лет и у 59% в возрасте от 6 до 10 лет. В дальнейшем этот процент снижается, поскольку поражение сердца прогрессирует и дети начинают предъявлять кардиальные жалобы [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
Патогенез поражения мышцы сердца при прогрессирующей мышечной дистрофии Дюшенна в настоящее время представляется следующим образом [Ishikawa Y, Bach JR, Sarma RJ et al., 1995]: прогрессирующая атрофия кардиомиоцитов и замещение их фиброзной тканью приводят к истончению миокарда (особенно левого желудочка, на который приходится основная гемодинамическая нагрузка), а также к снижению его способности к систолическому сокращению и диастолическому расслаблению. Выраженный фиброз в области задних папиллярных мышц ведет к пролабированию створок митрального клапана в полость левого предсердия (пролапс митрального клапана) с или без наличия митральной регургитации. Частота выявления пролапса митрального клапана у пациентов с прогрессирующей мышечной дистрофии Дюшенна составляет от 25 до 55% [Ishikawa Y, Bach JR, Sarma RJ et al., 1995]. Увеличение размеров левого предсердия как правило вторично, вследствие митральной регургитации или снижения сократительной способности левого желудочка. Нарушения ритма сердца и проводимости возникают вследствие прогрессирующего фиброза проводящей системы сердца [Novakovic I., Todorovic S., Apostolski S. et al., 1998].
Обычно поражение сердечной мышцы впервые диагностируется между 6 и 7 годами. С возрастом частота выявления кардиальных симптомов возрастает, и к 20 годам патология сердечно- сосудистой системы встречается у 95% больных. Наиболее частыми нарушениями, наблюдавшимися у 54% пациентов, были: тахикардия, аритмии и сердечная недостаточность. Особенно выражены данные симптомы в конечных стадиях заболевания [Adzija D et al.,1994].
Учитывая особенности двигательной активности пациентов с прогрессирующей мышечной дистрофией Дюшенна (а также с миодистрофией Бекера), отсутствие кардиальных жалоб, весьма малую физическую активность (как правило, больные к 10-11 годам теряют способность к самостоятельной ходьбе), часто обездвиженность на поздних этапах заболевания, G. Nigro с соавторами в 1993 году был предложен и введен в медицинскую практику новый диагностический термин – латентная сердечная недостаточность [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
Так же особенностями этой формы прогрессирующей мышечной дистрофии являются сопутствующая поражению мышц умственная отсталость, снижение интеллекта, остеопороз и истончение кортикального вещества костей, кардиомиопатия, легочно-сердечная недостаточность. У части больных обнаруживаются различные признаки эндокрпинопатии: адипозогенитальный синдром, низкорослость. В связи с дефицитом церебральных изоформ дистрофина — аподистрофинов, у 30 % больных с миодистрофией Дюшенна имеет место умственная отсталость различной степени: от пограничной интеллектуальной недостаточности до выраженной олигофрении. Тяжесть олигофрении и нарушений высших когнитивных функций не коррелирует с выраженностью мышечного дефекта и стадией миодистрофического процесса. К экзогенным факторам, усугубляющим проявления умственной отсталости, относят развивающуюся социальную дезадаптацию вследствие невозможности из-за двигательного дефекта полноценного участия детей в детских коллективах (сад, школа), влияние неблагоприятных перинатальных причин и, возможно, дисгенезий головного мозга (при КТ и МРТ изредка обнаруживают признаки церебральной атрофии) [Гринио Л.П., 1998].
Сопутствующие нарушения. Сухожильные и мышечные контрактуры (в том числе ахилловых сухожилий), прогрессирующий кифосколиоз, нарушение функции легких, кардиомиопатия, интеллект снижен. Мышечная слабость сочетается с пальпаторно определяемым увеличением и плотностью некоторых мышц (например, икроножных), что вначале является результатом гипертрофии, а затем замещения мышц жировой и соединительной тканью [Гринио Л.П., 1998].
Слабость дыхательной мускулатуры и диафрагмы обусловливает уменьшение ЖЕЛ до 20 % от нормы, что приводит к эпизодам ночной гиповентиляции. Дети часто встают со страхом, связанным с ощущением удушья, и боятся спать. Существенный вклад в летальность вносит дыхательная недостаточность, которая провоцируется интеркуррентными инфекциями или аспирацией [Тетенев Ф.Ф., Бодрова Т.Н., Емельянова Н.В., 2000].
Псевдогипертрофическая доброкачественная миодистрофия Беккера — Кинера.
Второй по частоте Х-сцепленной формой является так называемая доброкачественная форма псевдогипертрофической миодистрофии Беккера — Кинера. Впервые доброкачественная форма была описана в 1955 г. P . Becker и F . Kiener . В последующем V. McKu — sick (1964), R . Shaw , F . Dreifuss (1969) сообщили о подобном заболевании как самостоятельной мутации [Евтушенко С.К., Садеков И.А. 1994].
Менее тяжелая и реже встречающаяся, чем дистрофия Дюшенна, с более медленным течением и более поздним началом, но со сходными клиническими и лабораторными признаками. Это заболевание также является результатом дефекта в дистрофин-гене. Начинается в возрасте 10—15 лет. Поражаются мышцы проксимальных отделов конечностей, тазового и плечевого пояса [Bushby K.M.D. et al, 1993].
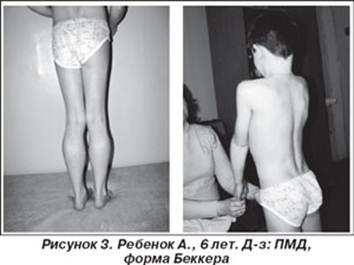
Течение сравнительно мягкое, длительное время сохраняется возможность самообслуживания и даже трудоспособность. В отличие от формы Дюшенна не наблюдается нарушений интеллекта. По топографии мышечного поражения миодистрофия Беккера—Кинера полностью повторяет миодистрофию Дюшенна. Слабость и гипотрофия вначале проявляются в мышцах тазового пояса и бедер, затем процесс распространяется на мышцы плечевого пояса. Мышцы лица обычно интактны. Псевдогипертрофии развиваются в икроножных мышцах почти во всех случаях, могут быть очень значительными, иногда они отмечаются и в других мышечных группах. Наблюдается прогрессирующий поясничный лордоз, появляется утиная походка, затруднение при подъеме с пола ("приемы миопата"), беге, а в поздних стадиях и при ходьбе. Развиваются сухожильные ретракции, в первую очередь в ахилловых сухожилиях. Кардиомиопатня при форме Беккера—Кинера или совсем не встречается или выражена очень слабо. При миодистрофий Беккера, как и при форме Дюшенна, повышен уровень креатинфосфокиназной активности, однако незначительно [Le-Thiet-Thanh; Nguyen-Thi-Man; Hori-S; Sewry-CA; Dubowitz-V, 1995].
До настоящего времени окончательно не решен вопрос о том, являются ли обе формы миодистрофий, сцепленные с Х-хромосомой, самостоятельными нозологическими формами или это разновидности течения одной болезни. Следует учесть, что при миодистрофий Беккера имеется сцепление с цветовой слепотой (иногда лишь частичной), что не встречается при форме Дюшенна. Не наблюдается благоприятного течения при экспериментальной миодистрофий у животных [Khurana T.S., Prendergast R.A., Alameddine H. et al., 1995]. С практической точки зрения, учитывая различный прогноз, следует обязательно различать эти две формы [Maeda M; Nakao S; Miyazato H; et al., 1996].
Электромиографические, биохимические и патоморфологические изменения умеренно выражены, отмечается изменение цветового зрения. Длительное время у больных сохраняются трудоспособность, возможность самостоятельного передвижения, интеллект; кардиомиопатия выражена умеренно [Maeda M; Nakao S; Miyazato H; et al., 1996].
Миодистрофия Дрейфуса — Хогана.
Мышечная дистрофия Дрейфуса— Хогана известна с конца 1960-х годов, когда была описана первая семья с Х-сцепленной формой болезни. Позднее наряду с другими описаниями этой формы появились единичные сообщения о клинически неотличимой миодистрофии с аутосомно-доминантным типом наследования. На протяжении нескольких десятилетий оба варианта, особенно аутосомно-доминантный, считались очень редкими. С развитием ДНК-диагностики эти представления изменились. Оказалось, что мышечная дистрофия Дрейфуса-Хогана вносит значимый вклад в структуру мышечных дистрофий. Относится к редким Х-хромосомным формам прогрессирующих мышечных дистрофий. Мышечная дистрофия Дрейфуса- Хогана является медленно прогрессирующей формой миодистрофии с Х-сцепленным рецессивным типом наследования [Белозеров Ю.М., Никанорова М.Ю., Перминов В.С., Страхова О.С., 2001].
Заболевание дебютирует между 5 и 15 годами жизни. Самыми ранними и типичными признаками обычно являются развивающиеся сгибательные контрактуры в локтевых суставах и разгибателях кистей, ретракции пяточных сухожилий. Затем возникает слабость и атрофия двуглавых и трехглавых мышц плеча, позже — дельтовидных мышц и других мышц плечевого пояса. В некоторых случаях в качестве первого симптома отмечают ходьбу на пальцах и наружных краях стоп, которая развивается приблизительно в 5-летнем возрасте. До этого момента двигательное развитие детей обычно адекватное. Мышечная слабость возникает незаметно и медленно прогрессирует [Мальмберг С.А., Петрухин А. С., Широкова В.И., 2000]. Примерно в 20-летнем возрасте наступает относительная стабилизация. Возможность ходьбы и подъема по лестнице сохраняется. Лицевая мускулатура остается интактной. Обычно имеется проксимальная слабость (лопаточно-плечевая) в руках и дистальная (перонеальная) в ногах. Приемы Говерса могут отсутствовать, сухожильные рефлексы не вызываются. Псевдогипертрофия икроножных мышц не характерна. Часто обнаруживается укорочение заднешейных мышц, ведущее к недостаточной подвижности шейного отдела позвоночника. Иногда встречается сколиоз вследствие уплотнения и, возможно, ретракции паравертебральных мышц, который с возрастом не нарастает. Характерен проксимальный тетрапарез [Карпович Е.И., Казакова Л.В., Колбасова Л.В. и др., 1998].
Частыми и прогностически важными признаками болезни являются нарушения сердечной проводимости и развивающаяся дилатационная или гипертрофическая кардиомиопатия. Последняя может осложняться развитием паралича предсердий вследствие фиброза импульсгенерирующих синусоатриальных клеток. В этих случаях показана имплантация искусственного водителя ритма. Синкопальные состояния и приступы брадикардии в некоторых случаях могут предшествовать появлению мышечной слабости, но чаще возникают на 3-м десятилетии жизни. Изменения в проводящей системе сердца далеко не всегда обнаруживают при стандартном ЭКГ-исследовании. Однако атриовентрикулярные блокады и периоды Венкебаха могут быть выявлены при 24-часовом холтеровском мониторировании. Аритмия, которую не удается устранить при имплантации искусственного водителя ритма, может привести к инсульту и смерти больного. Витальный прогноз при миодистрофии Дрейфуса – Хогана всецело зависит от степени поражения сердца [Темин П.А., Белозеров Ю.М., Никанорова М.Ю., Страхова О. С., 1998].
Юношеская псевдогипертрофия Мэбри
Первые симптомы появляются в пубертатном периоде (11—13 лет) в виде слабости в мышцах бедер и тазового пояса. Характерны выраженные псевдогипертрофии мышц, умеренный проксимальный тетрапарез. Сухожильные ретракции нетипичны. Интеллект сохранен, отсутствуют ретракции и контрактуры. Облигатным признаком является кардиомиопатия [Palmucci L., Doriguzzi C., Mongini T. et al., 2004].
Миодистрофия Роттауфа — Мортье
Начинается в возрасте 8—9 лет, отличается выраженным миосклерозом, характерной чертой болезни являются ранние, выраженные и быстропрогрессирующие сухожильные ретракции и контрактуры в локтевых, голеностопных суставах, ригидности позвоночника. Вначале мышечные атрофии развиваются в тазовом и плечевом поясах, проксимальных отделах конечностей и мышцах спины. Затем мышечные атрофии преобладают в лопаточно-плечевой области и в дистальных отделах конечностей. Из-за контрактур формируется ходьба на носках, а затем невозможность сгибания позвоночника вследствие фиброза мышц. Парезы мышц выражены умеренно и в основном затрагивают плечевой пояс и дистальные отделы ног. Псевдогипертрофии отсутствуют, интеллект сохранен. Течение заболевания медленное, больные длительно сохраняют подвижность и обслуживают себя. Характерна кардиомиопатия с нарушением проводящей системы сердца. К 35—40 годам может развиться полная атриовентрикулярная блокада, что определяет летальный исход. Содержание КФК значительно повышено и снижается в далеко зашедших стадиях процесса. Гетерозиготные носительницы здоровы, а уровень КФК у них нормальный. Клинические проявления близки миодистрофии Эмери—Дрейфуса, однако отмечается более диффузное распределение мышечных гипотрофии и большая скорость прогрессирования миодистрофического процесса [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].

Поясно-конечностная юношеская миодистрофия Эрба — Рота.
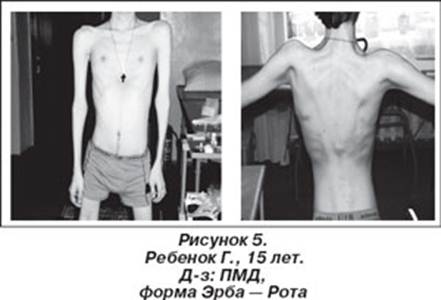
Первое сообщение о прогрессирующей мышечной дистрофии было опубликовано в России в 1895 г. врачом В.К. Ротом, который назвал заболевание мышечной сухоткой. Термин "конечностно-поясная мышечная дистрофия" употребляется для обозначения случаев проксимальной мышечной слабости, которая начинает развиваться на 2-м или 3-м десятилетии жизни, прогрессирует медленно и приводит к глубокой инвалидизации лишь через 15—20 лет. Представляет собой группу заболеваний, ключевым симптомом которых является слабость проксимальных мышц верхних и нижних конечностей. В некоторых случаях плечевой и тазовый пояса вовлекаются одновременно. Как правило, описывается наследственная аутосомно-рецессивная форма прогрессирующих мышечных дистрофий Эрба - Рота восходящего типа. Это заболевание наблюдается примерно в 30% случаев прогрессирующей мышечной дистрофии и представляет собой гетерогенную группу, объединенную по принципу локализации патологического процесса преимущественно в мышцах плечевого и тазового поясов. В тех случаях, когда начинается процесс атрофии мышц верхних конечностей и затем атрофируются мышцы тазового пояса, а через 2-4 года, как правило, развивается атрофия мышц нижних конечностей, констатируют нисходящий тип болезни. Имеются также спорадические случаи. Заболевание проявляется в возрасте 13—16 лет, однако первые его признаки могут наблюдаться в раннем детском возрасте (дистрофия Лейдена) [Мальмберг С.А. и соавт., 2001]. Характерными симптомами являются слабость и атрофия мышц тазового пояса и бедер, мышц живота и туловища, что проявляется гиперлордозом позвоночника, выпячиванием живота, утиной походкой, затруднением при переходе из горизонтального положения в вертикальное. Генерализация атрофий происходит по восходящему типу. Псевдогипертрофии икроножных мышц, ретракции и контрактуры выражены умеренно, интеллект сохранен. Больной стоит, несколько расставив ноги, чуть-чуть согнув их в коленях. Поясница сильно вогнута, живот выпячен, верхняя часть туловища откинута назад. При спокойно висящих верхних конечностях локти расположены позади туловища. Наибольшая выпуклость позвоночника находится на уровне 2-3 грудных позвонков, а наибольшая вогнутость соответствует верхним поясничным позвонкам. Лопатки немного приподняты, их внутренние края параллельны и сближены до расстояния 8 см, но значительно отстоят от грудной клетки. Плечи тонки, как у малого ребенка. Предплечья сравнительно с плечами кажутся нормальными. Ягодицы очень похудели. Бедра при сдвинутых ногах не касаются друг друга, они имеют цилиндрическую форму вследствие преобладающей атрофии приводящих мышц. Несмотря на значительное уменьшение объема многих мышц, нет ни одной парализованной. Чтобы переменить положение, например, лежачее на сидячее, больной должен повернуться спиной кверху, принять положение a la vache, опуститься ягодицами на пятки, затем разогнуть туловище, опираясь руками о постель, наконец, высвободить из под себя ноги. Чтобы встать с постели, больному приходится повернуться спиной кверху при помощи ряда окольных движений и ухваток, спустив потом одну за другой ноги на пол, а затем притупить к самому трудному маневру - разгибанию туловища. Для этого больной отыскивает руками более высокую точку опоры: стол, спинку кровати и т.п. и пользуется ею, чтобы поднять туловище на сколько можно, вслед за тем, отталкивая его рукой, а также брюшных мышц одной стороны, он достигает того, что туловище описывает дугу и перегибается на сторону. Когда туловище придет таким образом в одну фронтальную плоскость с нижними конечностями, уже небольшого напряжения мышц достаточно, чтобы отклонить его кзади. Спина принимает единственное положение, в котором больной может стоять без поддержки и ходить. Экспрессивность мутантных генов варьирует, что определяет существование тяжелых, легких и даже субклинических форм поясно-конечностной миодистрофии Эрба — Рота. Кардиомиопатия проявляется в поздних стадиях заболевания [Мальмберг С.А. и соавт., 2001]. Атрофия дыхательных мышц, деформация грудной клетки и позвоночника приводят к нарушению функции внешнего дыхания, легочно-сердечной недостаточности Смерть больных обычно наступает от легочных осложнения, в частности, бронхопневмонии [Тетенев Ф.Ф., Бодрова Т.Н., Емельянова Н.В., 2000].
Плече-лопаточно-лицевая миодистрофия Ландузи — Дежерина.
Тип наследования аутосомно-доминантный, имеются спорадические случаи. Обычно медленно прогрессирующее, умеренной тяжести заболевание. Заболевание начинается в возрасте 12—20 лет. Семейная отягощенность может быть не обнаружена, поскольку пораженные члены семьи зачастую не подозревают о своих собственных проблемах. Первоначально атрофии наблюдаются в плечевом поясе с последующим распространением на лицо, следствием чего являются амимия. Слабость мимической мускулатуры выражается в неспособности свистеть и потере выразительности лица. Типичны «полированный» лоб, лагофтальм, «поперечная» улыбка, толстые, иногда вывороченные губы (губы тапира). Тип течения болезни в большинстве случаев относительно благоприятный. Однако физические перегрузки, интенсивные спортивные занятия и нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни. Многие больные не становятся инвалидами и качество их жизни не ухудшается. Других больных приковывает к креслу-каталке в зрелом возрасте. Как правило, больные отмечают изменение своей мимики: их речь становится неразборчивой. На высоте заболевания грубо страдают круговые мышцы рта и глаза, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая и трехглавая мышцы плеча. Отмечаются характерные симптомы в виде поперечной улыбки (улыбки Джоконды), протрузии верхней губы (губы тапира). Грудная клетка уплощается в переднезаднем направлении, плечевые суставы ротируют внутрь, лопатки приобретают крыловидную форму при попытке поднять руки вверх, деформация грудной клетки и позвоночника, скошенные плечи, появление широкого межлопаточного промежутка, уплощение грудной клетки, сколиоз. Атрофии распространяются в нисходящем направлении, и в процесс вовлекаются мышцы ног (лопаточно-плечебедренный, лицелопаточно-плечеперонеальный. лицелопаточно-плечеягодично-бедренный, лицелопаточно-плече-ягодично-бедренно-перонеальный и другие варианты). В таких случаях слабость наиболее заметна в группе малоберцовых мышц по свисающей стопе, но может быть и в проксимальных отделах ног [Бадалян Л.О., 2008]. Генерализация патологического процесса продолжается 10—15 лет, он постепенно распространяется на мышцы тазового пояса, проксимальных и дистальных отделов ног. Свисание стоп и слабость ног может вызывать падения больного и прогрессирующее затруднение движений. В некоторых случаях развиваются также атрофии мышц бедер и голеней. Псевдогипертрофия икроножных, дельтовидных, лицевых мышц выражена умеренно. Рефлексы могут быть долгое время сохранены. Характерной клинической особенностью является асимметрия атрофии. Возможно некоторое обратное развитие симптомов. Могут наблюдаться псевдогипертрофии мышц. Контрактуры и ретракции выражены умеренно. Кардиомиопатия редка. Аномалии сосудов сетчатки, которые могут быть обнаружены у многих больных при использовании метода ангиоретинмографии, рассматриваются в качестве составляющей части фенотипических проявлений болезни. В большинстве случаев с тяжелыми глазными проявлениями находят телеангиэктазии, отек и отслойку сетчатки. Может наблюдаться также снижение слуха. При выявлении телеангиэктазии их ликвидируют с помощью коагуляции, что предотвращает развитие слепоты. Больные длительное время сохраняют трудоспособность. Псевдогипертрофии выражены в икроножных и дельтовидных мышцах. Мышечный тонус в ранних стадиях болезни снижен в проксимальных группах мышц. Глубокие рефлексы снижены преимущественно с двуглавой и трехглавой мышц плеча. Интенсивные физические нагрузки ведут к быстрому прогрессированию заболевания [Сухомясова А.Л., 2005].
Лопаточно-перонеальная дистрофия.
Болезнь наследуется по сцепленному с Х-хромосомой типу и может являться аллельным вариантом для миодистрофии Эмери—Дрейфуса. До 5-летнего возраста дети здоровы, однако затем начинается деградация психики, которая проявляется неспособностью к обучению и отставанием умственного развития. Вскоре возникают слабость и атрофии лопаточных или плечевых и малоберцовых мышц. Контрактуры и псевдогипертрофии мышц не развиваются. Симптомы кардиомиопатии наблюдаются в подростковом возрасте и определяют летальный исход. Клиническая картина такая же, как при плечелопаточно-лицевой дистрофии, но нет мышечной слабости лица, возможны явления кардиомиопатии. В большинстве случаев заболевание начинается в среднем возрасте и наследуется по аутосомно-доминантному типу, но может встречаться и форма болезни с ранним началом, другим механизмом генетической передачи (связанный с Х-хромосомой, рецессивный), с клиническими проявлениями суставных контрактур и кардиомиопатией (тип Эмери—Дрейфуса) [Palmucci L., Doriguzzi C., Mongini T. et al., 2004].
Дистальная миодистрофия Говерса

Относится к редким заболеваниям, характеризуется генетической гетерогенностью. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Проявляется в возрасте 30—60 лет. Симптоматика поражения мышц дистальных отделов конечностей. Характерными симптомами являются слабость и атрофии мышц голени и стоп, снижение ахилловых и коленных рефлексов. Ведущими симптомами являются шлепающие стопы, слабость мышц — разгибателей кисти. Вначале слабость и атрофия имеют место в икроножных мышцах, передняя группа мышц голеней остается интактной. Генерализация процесса с развитием атрофий кистей и проксимальных отделов конечностей происходит в течение 5—10 лет. Ахилловы рефлексы отсутствуют, все остальные вызываются [Бадалян Л.О., 2008]. У больных, как правило, выражена кардиомиопатия, приводящая к летальному исходу. Течение: доброкачественное, медленно прогрессирующее [Piantadosi C., Nigro V., Servider S. et al., 1998].
Офтальмоплегическая и окулофарингеальная (глазо-глоточная) формы.
Впервые заболевание было описано von Graefe в 1868 г. как «прогрессирующая наружная офтальмоплегия», в 1915 г. — Тейлором как семейный случай комбинации птоза век и паралича глотательных мышц. Однако только в 1962 г. М. Victor связал «прогрессирующую наружную офтальмоплегию» с фарингеальной слабостью и дал заболеванию название «окулофарингеальная миодистрофия» [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Генетически обусловленные миодистрофии глазных мышц делятся на несколько форм:
I. Изолированная окулярная миодистрофия, начинающаяся в молодом возрасте и приводящая к полной наружной офтальмоплегии, обычно без явлений диплопии. В ряде случаев процесс распространяется на другие поперечно-полосатые мышцы. Повышается уровень креатинфосфокиназы, лактатдегидрогеназы и альдолазы в сыворотке крови. Аутосомно-доминантный тип наследования.
II. Поздняя окулярная миодистрофия наблюдается в виде следующих форм: окулофарингеальная - поражение глазодвигательных и глоточных мышц с нарушением глотания; окулофациальная форма (отличается от формы Ландузи-Дежерина только сохранностью функции круговой мышцы глаза); окулобрахиальная форма (сочетается с поражением мышц проксимальных отделов конечностей); окулокардиальная форма.
III. Окулярная миодистрофия, сочетающаяся с немышечными поражениями дегенеративного характера, проявляющаяся задержкой развития пирамидной системы, морфологической или функциональной недостаточностью половых желез, сердечной деятельности. Это так называемый синдром Кирнса- Сейра - к офтальмоплегии присоединяется поражение центральной и периферической нервной системы (окуло-краниосоматическое нервно-мышечное заболевание). Появляется у детей и подростков с низким ростом, недостаточностью умственного развития, атаксией, глухотой, пигментной ретинопатией, дефектами проводящей системы сердца. Передается по аутосомно-доминантному и аутосомно-рецессивному типу [Becher M. W., Morrison L., Davis L.E. et al., 2001].
Тип наследования аутосомно-доминантный, имеются спорадические случаи. Возраст начала заболевания может быть разным. Чаще начало заболевания в возрасте 50-60 лет с явлениями птоза, ограничения экстраокулярных движений, лицевой и крикофарингеальной мышечной слабости [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
В клинической картине заболевания наблюдается прогрессирующая мышечная слабость и атрофия проксимальных отделов конечностей, расстройства глотания и фонации, птоз, слабость лицевой мускулатуры. Характерны медленно нарастающая слабость и атрофия глазодвигательных мышц, ограничение движений глазных яблок. Крикофарингеальная мышечная слабость ведет к ахалазии, дисфагии и аспирации. Так как нарушения движений глаз носят хронический характер, они редко ведут к диплопии.
Поражение глазных мышц может быть изолированным или сочетаться с атрофией мышц лица, глотки (что приводит к затруднению глотания), поражением мышц конечностей. Течение медленно прогрессирующее [Blumen S.C., Brais B., Korczyn A.D. et al., 1999].
Миодистрофия Бетлема.
Дебют: раннее детство. Заболевание начинается со слабости мышц тазового пояса, которая возникает в грудном или раннем детском возрасте. Лицевая мускулатура остается интактной. Часто симптомы болезни настолько стертые, что родственники остаются неосведомленными об имеющихся отклонениях. Слабость прогрессирует медленно и обычно не приводит к инвалидизации и не влияет на продолжительность жизни. Рано развиваются сгибательные контрактуры в локтевых, голеностопных и межфаланговых суставах (кроме больших пальцев). Деформаций позвоночника не наблюдается. Ретракция пяточных сухожилий является причиной ходьбы на пальцах. Сухожильные рефлексы нормальны или снижены. Кардиомиопатия нехарактерна. Течение: доброкачественное, стационарное [Piantadosi C., Nigro V., Servider S. et al., 1998].

Похожие работы
... Hungary. – CD. – A0034. Автор провів аналіз клініко-ортопедичних даних у хворих на ПМД, який було покладено в основу написання тезів. АНОТАЦІЯ Зима А.М. Діагностика та ортопедичне лікування різних форм прогресуючої м’язової дистрофії. Рукопис. Дисертація на здобуття наукового ступеня кандидата медичних наук за спеціальністю 14.01.21 – травматологія та ортопедія ДУ «Інститут травматології та ...
... ревматизма обусловила значительное снижение заболеваемости — до 0Д8 на 1000 детского населения. В разработку проблемы детского ревматизма внесли большой вклад отечественные педиатры В. И. Молчанов, А. А. Кисель, М. А, Скворцов, А. Б. Воловик, В. П. Бисярина, А. В. Долгополова и др. Эпидемиология, Установлена связь между началом заболевания и перенесенной стрептококковой инфекцией, в основном в ...
... 036.При инфаркте в бассейне передней артерии сосудистого сплетения (передняя ворсинчатая) не бывает #а)гемиплегии #б)гемианестезии *#в)афазии #г)вазомоторных нарушений в области парализованных конечностей #д)гемианопсии 037.Препараты наперстянки и строфанта при декомпенсации дисциркуляторной энцефалопатии назначают #а)для нормализации сердечного ритма ...
... КМГ, которая в начале может быть парциальной. Размеры сердца и степень увеличения отдельных камер в большой сетпени зависят от характера порока. См. Методическое пособие "Дифференциальный диагноз при шумах сердца". Синдром Марфана. Комплекс наследственных аномалий (наследование аутосомно-доминантное), связанных с поражением соединительной ткани. Типичны изменения скелета, включащие ненормально ...
0 комментариев