Навигация
Уменьшение погрешности определения в 2-3 раза за счет увеличения числа измерений в одной пробе
14779
знаков
4
таблицы
3
изображения
1. Уменьшение погрешности определения в 2-3 раза за счет увеличения числа измерений в одной пробе.
2. Метод добавок не требует тщательной стабилизации ионной силы в анализируемой пробе, так как ее колебания отражаются на величине абсолютного значения потенциала в большей степени, чем на величине наклона электродной функции. В связи с этим погрешность определения по сравнению с методом градуировочного графика уменьшается.
3. Применение целого ряда электродов проблематично, так как наличие недостаточно стабильного потенциала требует частого проведения процедуры градуировки. Поскольку в большинстве случаев дрейф потенциала мало сказывается на наклоне калибровочной функции, то получение результатов методом стандартных добавок и методом Грана существенно повышает точность и упрощает процедуру анализа.
4. Метод стандартных добавок позволяет контролировать правильность проведения каждого аналитического определения. Контроль производится во время обработки экспериментальных данных. Так как в математической обработке принимает участие несколько экспериментальных точек, то проведение через них прямой каждый раз подтверждает то, что математический вид и величина наклона калибровочной функции не изменились. В противном случае линейный вид графика не гарантирован. Таким образом, возможность контроля правильности анализа в каждом определении повышает надежность получения результатов.
Как уже отмечалось, метод стандартных добавок позволяет проводить определения в 2-3 раза точнее, чем метод градуировочного графика. Но для получения такой точности определения следует пользоваться одним правилом. Чрезмерно большие или малые добавки снижают точность определения. Оптимальная величина добавки должна быть такой, чтобы она вызывала отклик потенциала в 10-20 мВ для однозарядного иона. Это правило оптимизируют случайную погрешность анализа, однако в тех условиях, в которых часто применяется метод добавок, становится значимой систематическая погрешность, связанная с изменением характеристик ионоселективных электродов. Систематическую погрешность в этом случае полностью определяет погрешность от изменения наклона электродной функции. Если в течение эксперимента изменился наклон, то при определенных условиях относительная погрешность определения будет приблизительно равна относительной погрешности от изменения наклона.
2. Метод двойной стандартной добавки.
Метод заключается в том, что к анализируемому раствору добавляются 2 порции стандартного раствора. Величина этих порций одинакова. По результатам измерений вычисляется параметр
R = D E2 / D E1 , где
D E1 - разность между потенциалом электродов в анализируемом растворе, и в растворе после первой добавки; D E2 - разность между потенциалом электродов в анализируемом растворе, и в растворе после второй добавки.
Пользуясь вычисленным параметром, по специальной таблице находится искомое значение концентрации. Использование таблицы оправдано тем, что для поиска значения концентрации приходится решать трансцендентное уравнение
R = lg(1/(1+2 DV/W) + 2DC/Cx) / lg(1/(1+DV/W) + DC/Cx) .
Следует уточнить, что DС - концентрация в анализируемом растворе после добавки, если бы в этом растворе не было бы больше никаких ионов, т.е. DС = Сисх DV / (W+ DV). Сисх - концентрация в стандартном растворе.
Решать трансцендентное уравнение, каждый раз, когда это нужно, затруднительно, поэтому лучше пользоваться следующей таблицей.
| DV/W | 0,001 | 0,01 | 0,02 | 0,04 | 0,06 | 0,08 | 0,1 | 0,15 | 0,2 |
| DC/Cx | параметр R | ||||||||
| 0,1 | 1,9137 | 1,9224 | 1,9366 | 1,9861 | 2,0902 | 2,3527 | 3,6233 | 1,0112 | 1,2989 |
| 0,2 | 1,8461 | 1,8523 | 1,8608 | 1,8839 | 1,9168 | 1,9624 | 2,0255 | 2,3248 | 3,3002 |
| 0,3 | 1,7919 | 1,7968 | 1,8031 | 1,8190 | 1,8393 | 1,8648 | 1,8961 | 2,0063 | 2,1835 |
| 0,4 | 1,7473 | 1,7513 | 1,7563 | 1,7684 | 1,7833 | 1,8009 | 1,8216 | 1,8879 | 1,9786 |
| 0,5 | 1,7099 | 1,7132 | 1,7174 | 1,7271 | 1,7387 | 1,7521 | 1,7674 | 1,8142 | 1,8736 |
| 0,6 | 1,6779 | 1,6807 | 1,6842 | 1,6923 | 1,7017 | 1,7125 | 1,7246 | 1,7603 | 1,8037 |
| 0,7 | 1,6501 | 1,6526 | 1,6556 | 1,6625 | 1,6704 | 1,6793 | 1,6892 | 1,7178 | 1,7516 |
| 0,8 | 1,6258 | 1,6280 | 1,6306 | 1,6366 | 1,6433 | 1,6509 | 1,6592 | 1,6828 | 1,7103 |
| 0,9 | 1,6043 | 1,6063 | 1,6086 | 1,6138 | 1,6197 | 1,6262 | 1,6333 | 1,6533 | 1,6762 |
| 1 | 1,5851 | 1,5869 | 1,5889 | 1,5935 | 1,5987 | 1,6044 | 1,6106 | 1,6279 | 1,6474 |
| 1,1 | 1,5679 | 1,5694 | 1,5713 | 1,5754 | 1,5800 | 1,5851 | 1,5905 | 1,6056 | 1,6225 |
| 1,2 | 1,5523 | 1,5537 | 1,5553 | 1,5591 | 1,5632 | 1,5677 | 1,5725 | 1,5859 | 1,6007 |
| 1,3 | 1,5380 | 1,5393 | 1,5408 | 1,5442 | 1,5479 | 1,5520 | 1,5563 | 1,5683 | 1,5815 |
| 1,4 | 1,5250 | 1,5262 | 1,5276 | 1,5307 | 1,5341 | 1,5377 | 1,5417 | 1,5524 | 1,5642 |
| 1,5 | 1,5131 | 1,5141 | 1,5154 | 1,5182 | 1,5213 | 1,5247 | 1,5283 | 1,5380 | 1,5487 |
| 1,6 | 1,5020 | 1,5030 | 1,5042 | 1,5068 | 1,5096 | 1,5127 | 1,5160 | 1,5249 | 1,5346 |
| 1,7 | 1,4918 | 1,4927 | 1,4938 | 1,4962 | 1,4988 | 1,5017 | 1,5047 | 1,5128 | 1,5217 |
| 1,8 | 1,4822 | 1,4831 | 1,4841 | 1,4864 | 1,4888 | 1,4914 | 1,4942 | 1,5017 | 1,5099 |
| 1,9 | 1,4734 | 1,4742 | 1,4751 | 1,4772 | 1,4795 | 1,4819 | 1,4845 | 1,4915 | 1,4990 |
| 2 | 1,4651 | 1,4658 | 1,4667 | 1,4686 | 1,4708 | 1,4730 | 1,4754 | 1,4819 | 1,4889 |
| 2,1 | 1,4573 | 1,4580 | 1,4588 | 1,4606 | 1,4626 | 1,4647 | 1,4670 | 1,4730 | 1,4795 |
| 2,2 | 1,4499 | 1,4506 | 1,4514 | 1,4531 | 1,4549 | 1,4569 | 1,4591 | 1,4647 | 1,4708 |
| 2,3 | 1,4430 | 1,4436 | 1,4444 | 1,4460 | 1,4477 | 1,4496 | 1,4516 | 1,4569 | 1,4626 |
| 2,4 | 1,4365 | 1,4371 | 1,4378 | 1,4393 | 1,4410 | 1,4427 | 1,4446 | 1,4496 | 1,4549 |
| 2,5 | 1,4303 | 1,4309 | 1,4315 | 1,4330 | 1,4345 | 1,4362 | 1,4380 | 1,4427 | 1,4477 |
| 2,6 | 1,4244 | 1,4250 | 1,4256 | 1,4270 | 1,4285 | 1,4300 | 1,4317 | 1,4362 | 1,4409 |
| 2,7 | 1,4189 | 1,4194 | 1,4200 | 1,4213 | 1,4227 | 1,4242 | 1,4258 | 1,4300 | 1,4345 |
| 2,8 | 1,4136 | 1,4141 | 1,4146 | 1,4159 | 1,4172 | 1,4186 | 1,4201 | 1,4241 | 1,4284 |
| 2,9 | 1,4085 | 1,4090 | 1,4095 | 1,4107 | 1,4120 | 1,4133 | 1,4148 | 1,4186 | 1,4226 |
| 3 | 1,4037 | 1,4042 | 1,4047 | 1,4058 | 1,4070 | 1,4083 | 1,4097 | 1,4133 | 1,4171 |
В изложенных выше формулах отсутствует упоминание о наклоне электродной функции, так как это было целью разработки метода двойной стандартной добавки. На первый взгляд это факт выгодно отличает этот метод, поскольку процедура анализа упрощается, но это не так. Выиграв в одном, мы, безусловно, теряем в другом. Во-первых, для того, чтобы ошибка анализа была удовлетворительной, нужно быть уверенным в линейности электродной функции ионоселективного электрода! Отклонения от линейности будут приводить к очень большой ошибке анализа. Таким образом калибровать электроды все равно придется. Во-вторых, случайная погрешность анализа существенно больше, чем в методе Грана и методе стандартной добавки. Например, ошибка измерения потенциала в 1мВ может приводить к погрешности анализа в 10-20%.
Обобщая вышесказанное, напрашивается вывод о том, что метод двойной стандартной добавки лучше применять только для очень надежных в эксплуатации электродов, таких как, например, фторидселективный.
3. Метод добавок в условиях нелинейной калибровки.
Изложенные выше различные варианты метода добавок имеют одно общее свойство, заключающееся в том, что в основе их лежит закон Нернста. Закон предполагает линейность электродной функции в неограниченном диапазоне концентраций анализируемого иона. Если электродная функция нелинейна, то применение известных методов добавок становится неоправданно рискованным. Что делать в этом случае?
При наличии под рукой компьютера и хорошего математика, можно прекрасно организовать анализ методом добавок по новому алгоритму. Отсутствие компьютера, а тем более математика, ставит личные аналитические планы под вопрос.
Есть способ решения этой задачи, насколько экстравагантный, настолько и практичный. Он состоит в следующем. Проводится обычная серия экспериментов, включающая в себя калибровку ионоселективного датчика и определения искомых концентраций в анализируемых пробах введением добавок стандартного раствора в пробу. Результаты измерений наносятся на миллиметровую бумагу в виде кривых. Координаты графиков - E (потенциал) от V (введенного объема добавок). Общий вид кривых показан на рис.3. Точки, принадлежащие одной кривой, объединяются графически сплошной линией с помощью лекала. Не стоит пытаться найти одну лекальную кривую. Лучше провести несколько кривых. Затем калибровка переносится на кальку, причем на кальке обозначаются оси координат. Кривая на кальке совмещается последовательно с кривыми на миллиметровой бумаге. При совмещении следует пользоваться следующим правилом: оси абсцисс на кальке и миллиметровой бумаге должны быть параллельны. Единственность такого совмещения обеспечивает кривизна графиков.
|
|
| Рис. 3 |
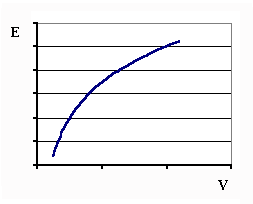
После того, как кривые будут совмещены, расстояние между осями ординат будет равно эквивалентному объему, пропорциональному искомой концентрации анализируемого иона.
Как видно из описания, метод несложен, но он позволяет с малыми затратами энергии работать и получать результаты в условиях нелинейной калибровки.
По сложившейся традиции рассмотрим приемы, которые оптимизируют процедуру анализа и снижают погрешность определения.
Перед тем, как работать в области нелинейности калибровки, надо быть уверенным в том, что такой вид калибровки вызван только реакцией потенциалопределяющего иона. Помимо потенциалопределяющего иона, нелинейность калибровки может вызываться режимом перемешивания пробы, растворимостью мембраны электрода и т.п. Перечисленные факторы плохо поддаются контролю, и будут вызывать, скорее всего, недостоверные результаты.
Величина первой добавки должна быть примерно равной тому количеству анализируемого иона, что уже есть в пробе. Слишком большие или слишком малые добавки неизбежно вызовут увеличение случайной ошибки определения.
При совмещении кривых следует отдавать предпочтение начальным точкам кривой, т.е. тем точкам, где динамика изменения потенциала от введенной добавки наибольшая.
Чем меньше объем вводимой добавки, тем лучше будут совпадать кривые. Для того чтобы оценить меру влияния объема пробы на точность анализа, были проведены модельные расчеты. Оказалось, что если отношение вводимого объема к общему объему пробы не превышает величины V/W=0,1, то погрешность анализа не будет превышать 1,5%. Эти расчеты верны только для линейной калибровки. Если есть отклонения от линейности, то отношение V/W должно быть меньше. Оценить точнее погрешность нельзя, так как расчеты будут зависеть от конкретного вида нелинейности калибровки.
В заключение следует привести значение погрешности, которое можно получить, используя новый метод. Расчеты показали, что по точности новый метод почти не уступает методу Грана, так как была достигнута погрешность расчетов в 1%.
Список литературы
Для подготовки данной работы были использованы материалы с сайта http://www.novedu.ru/
Похожие работы


... и природы вещества, участвующего в электрохимической реакции. Электрохимические параметры при этом служат аналитическими сигналами, при условии, что они измерены достаточно точно. Электрохимические методы анализа в практику химического анализа вошли сравнительно давно и занимают в ней важную роль. Впервые потенциометрическое титрование было проведено в 1893 г. в институте Оствальда в Лейпциге, а ...
... случае мосты обычно заполняют растворами различных солей с концентрациями не выше 1М. Вследствие этого возникает большая вероятность того, что потенциал электрода не будет стабилен. Систематический поиск неисправностей Неблагополучное проведение определения можно выявить как при измерении потенциала ионоселективного электрода, так и при вычислении результатов анализа.На этапе непосредственных ...
... модель параллельно соединенных источников э.д.с. В этой модели часть потенциалопределяющих реакций рассматриваются в виде источников э.д.с. в том виде, как это принято в электродинамике: где e 1 - э.д.с. источника тока, организованного потенциалопределяющим ионом; e 2 - э.д.с. источника тока, организованного альтернативной электрохимической реакцией; R1 и R2 - внутренние сопротивления ...

... , основанной на поглощении атомами рентгеновского излучения. Ультрафиолетовая спектрофотометрия — наиболее простой и широко применяемый в фармации абсорбционный метод анализа. Его используют на всех этапах фармацевтического анализа лекарственных препаратов (испытания подлинности, чистоты, количественное определение). Разработано большое число способов качественного и количественного анализа ...
0 комментариев