Навигация
Методы определения азота
118647
знаков
2
таблицы
3
изображения
2.3 Методы определения азота
Существующие методы определения содержания азота в сырье, полуфабрикатах и готовой продукции можно разделить на две группы: методы, предусматривающие сжигание (минерализацию) навески исследуемого продукта; методы, не предусматривающие сжигание навески.
В анализах, проводимых в лабораториях береговых рыбообрабатывающих предприятий и судов, наиболее часто используются методы, относящиеся к первой группе. Некоторые из них достаточно быстрые. Снижение затрат времени на анализ достигается за счет рационального подбора количественного и видового состава основных реагентов и катализаторов, а также совмещения отдельных операций (например, минерализации, отгонки и улавливания аммиака) и изменения техники их проведения (например, замена титрования спектрофотометрическим анализом).
В основе методов, не предусматривающих минерализацию навески, лежат цветные реакции, которые протекают в результате взаимодействия белков с некоторыми химическими реактивами.
Определение содержания общего азота (арбитражный метод). По этому методу общий азот должен быть определен в виде аммиака (NНз) после разрушения азотсодержащего вещества (продукта) горячей концентрированной H2SO4. Образовавшийся при разложении сульфат аммония [(NH4)2SO4] следует разрушить концентрированной щелочью, и полученный NH3 отогнать с паром в титрованный 0,1 н. раствор H2SO4. Определение закончить обычным ацидометрическим титрованием.
Навеску исследуемого продукта (мука в количестве 0,2...0,5 г, фарш — 0,5...1,0 г), отвешенную с погрешностью не более 0,0001 г, следует поместить в трубочку из фильтровальной бумаги или станиоля, закрытую с одной стороны. Диаметр ее должен быть несколько меньше диаметра горла колбы, в которой будет проводиться мокрое сжигание. Около 5 г тузлука (в зависимости от содержания в нем азота) осторожно влить в колбу на 100 см3, не касаясь стенок горла последней. Затем к навеске добавить несколько мелких кристаллов медного купороса (0,2...0,3 г) и прилить 10 см3 H2SO4, плотностью 1,84 г/см3. Колбу с содержимым осторожно нагреть в вытяжном шкафу, не допуская разбрызгивания жидкости.
Когда содержимое колбы сделается однородным, нагревание прекратить, дать остыть массе, прибавить 0,5 г сернокислого калия и снова нагревать до тех пор, пока жидкость в колбе не станет прозрачной, зеленовато-голубого цвета без бурого оттенка. Внутренние стенки колбы должны быть совершенно чистыми. Это достигается осторожным взбалтыванием содержимого колбы до смывания со стенок темных обугленных частиц муки.
По окончании сжигания содержимое колбы охладить и перенести в отгонную колбу на 500...750 см3. Колбу для сжигания необходимо тщательно сполоснуть, проверяя полноту смывания путем прибавления 1...2 капель раствора метилового красного. Для перенесения сожженной навески требуется 200...250 см3 дистиллированной воды. Приемником служит коническая колба на 250...300 см3, в которую предварительно должно быть налито 25...30 см3 0,1 н. раствора H2SO4. Конец трубки холодильника должен быть погружен в раствор H2SO4.
В отгонную колбу осторожно, по стенкам, избегая смешивания жидкостей, следует прилить 50...70 см3 33%-го раствора NaOH. В колбу бросить кусочек лакмусовой бумаги и быстро закрыть пробкой, соединенной каплеуловителем с холодильником. Осторожно перемешивая содержимое колбы, сразу же начинать ее нагревание. Реакция жидкости в колбе должна быть резко щелочной. После того как жидкость в колбе бурно закипит, приемник опустить с таким расчетом, чтобы конец трубки холодильника находился на некотором расстоянии от поверхности жидкости. В таком положении продолжать отгонку до тех пор, пока из колбы отгонится не менее 2/3 содержащейся в ней жидкости. Кроме того, конец отгонки определяют проверкой реакции дистиллята по лакмусовой бумаге. Если отгонка закончена, то капля дистиллята не должна вызывать посинения лакмусовой бумаги. В конце отгонки при кипении массы появляются характерные толчки, свидетельствующие о прекращении отгонки. По окончании отгонки конец трубки холодильника смыть водой в приемную колбу и содержащийся в приемнике избыток H2SO4 оттитровать 0,1 н. раствором едкой щелочи в присутствии метилового красного или двойного индикатора метилового красного или метилового синего.
Параллельно в тех же условиях, но без навески исследуемого вещества, провести контрольный опыт.
Содержание общего азота Х (в %) вычисляется по формуле:
x = (v-v1)*k*0.0014*100 / m
где v — объем 0,1 н. раствора едкой щелочи, пошедший на титрование H2SO4 в контрольном опыте, см3; v1 — объем 0,1 н. раствора едкой щелочи, пошедший на титрование избытка H2SO4 в рабочем опыте, см3; k — коэффициент пересчета на точно 0,1 н. раствор щелочи; 0,0014 — количество азота, эквивалентное 1 см3 0,1 н. раствора едкой щелочи; т — масса навески исследуемого продукта, г.
Расхождение между параллельными определениями не должно превышать 0,3%.
Количество белковых веществ определяется путем умножения азота на коэффициент, соответствующий данному продукту (например, для сырья, содержащего белки мышечных и нервной тканей — протамины, гистоны, альбумины, глобулины — 6,25; белки опорно-трофических и эпителиальных тканей — протеиноиды, альбуминоиды, склеропротеины — 5,71).
Полумикрометод определения содержания общего азота (стандартный метод). Минерализацию навески следует проводить так же, как по арбитражному методу. Массу навески увеличивают до 0,5 г, так как в дальнейшем проводится разведение.
Колориметрический метод определения содержания общего азота (нестандартный метод). Метод основан на способности NH-, давать интенсивное ярко-желтое окрашивание с реактивом Несслера.
Определение содержания белкового и небелкового азота. Исследуемый материал должен быть смешан с водой. К смеси следует добавить реактив, осаждающий белок. Выпавший осадок белка отфильтровать и определить содержание азота в осадке и в фильтрате. Азот осадка соответствует белковому азоту, а азот фильтрата — небелковому. Если известно содержание общего азота в исследуемом материале, можно ограничиться определением азота только в осадке или в фильтрате и по разности между общим азотом в исследуемом материале и азотом в осадке или в фильтрате вычислить количество белкового азота.
Метод определения содержания белкового азота основан на способности белковых веществ образовывать с гидратом окиси меди Сu(ОН)2 соединения, не растворимые даже в горячей воде. Количество азота в полученном осадке определяется арбитражным или другим стандартным методом.
Для определения содержания азота истинных белков (белковый азот) следует отвесить 0,5...1,0 г (с погрешностью не более 0,01 г) тонко растертого в ступке исследуемого материала и поместить его в термостойкий химический стакан на 100...150 см3. Добавить 50 см3 дистиллированной воды и нагреть до кипения. К нагретой массе (смеси) прилить 25 см3 раствора медного купороса (60 г CuSO4.5H2O растворить в 1000 см3 дистиллированной воды) и при постоянном помешивании прилить 25 см3 раствора NaOH (12,5 г NaOH растворить в 1000 см3 дистиллированной воды).
После отстаивания смеси жидкость осторожно слить декантацией через бумажный фильтр, а осадок в стакане промыть несколько раз горячей дистиллированной водой, сливая промывные воды через тот же фильтр. Промывание вести до тех пор, пока фильтрат не перестанет давать реакцию на H2SO4 (проба с хлористым барием). Промытый осадок количественно перенести на фильтр, просушить и вместе с фильтром сжечь в колбе для сжигания. Все дальнейшие операции, начиная с сжигания пробы, выполнять так же, как и при определении общего азота арбитражным или другим стандартным методом с использованием в процессе минерализации катализаторов или их смеси.
Параллельно провести контрольный опыт в тех же условиях, но без навески, что позволит установить содержание азота в фильтре и в реактивах. Результаты контрольного опыта учесть при расчете содержания общего азота в исследуемом материале. Содержание истинных белков определить путем умножения полученного количества азота на коэффициент 6,25.
При определении белкового азота в мясе жирных рыб собранный на фильтре осадок после высушивания следует промыть петролейным эфиром и снова подсушить. Удаление жира облегчает последующее сжигание осадка с фильтром.
Метод достаточно хорош, но не безупречен, так как Сu(ОН)2 осаждает частично пептоны. Кроме того, целый ряд аминокислот дает труднорастворимые медные соли, которые, попадая в белковый осадок, трудно вымываются, что способствует завышению результатов определения. При наличии в исследуемом материале лецитинов, азот последних также присоединяется к белковому азоту.
Определение содержания летучих оснований азота (АЛО). К летучим основаниям относится ряд соединений, в том числе NH3 монометиламины (CH3NH2), диметиламины [(СНз)2NН] и триметиламины [(CH3)3N или ТМА]. Количественное содержание АЛО является одним из объективных показателей свежести сырья и готовой продукции. Сущность метода состоит в том, что связанные и свободные летучие основания отгоняются паром, а затем отфильтровываются.
Навеску сухого продукта (например, муки) массой около 5 г или сырого (например, фарша) массой до 10 г (отвешенную с погрешностью не более 0,01 г) следует поместить в отгонную колбу на 500 см3. В колбу добавить 250 см3 дистиллированной воды, 25 см3 5%-го магнезиального молока или 1 г окиси магния (магнезии) — MgO и, во избежание вспенивания, кусочки чистого парафина. Содержимое колбы перемешать. Реакция смеси должна быть щелочной (контролировать по внесенной в колбу красной лакмусовой бумажке). Колбу закрыть пробкой, соединяющей ее с каплеуловителем. Приемником должна служить коническая колба на 300 см3, в которую предварительно следует налить 25 см3 0,1 н. раствора НС1. Через суспензию, содержащуюся в отгонной колбе, необходимо интенсивно пропускать пар из парообразователя. При этом отгонную колбу слабо подогревать. Конец холодильника в начале отгона должен быть опущен в раствор H2SO4. Когда объем (дистиллята) в приемной колбе достигнет 200...250 см3, отгон прекратить. Окончание отгона следует контролировать с помощью лакмусовой бумажки. При нанесении на бумагу капли дистиллята, выходящего из холодильника, реакция должна быть нейтральной. После прекращения отгона содержимое приемной колбы оттитровать 0,1 н. раствором NaOH в присутствии 3...4 капель индикатора метилрота (0,2%-ный раствор метилового красного в 60%-ном этиловом спирте).
Одновременно необходимо провести контрольный опыт. Все операции проводить так же, как и в стандартном опыте, но без навески исследуемого продукта.
Содержание АЛО на 100 г исследуемого продукта (мг%) вычисляется по формуле:
x = (v-v1)*k*1.14*100 / m
где v — объем 0,1 н. раствора NaOH, пошедший на титрование контрольной пробы, см3; v1— объем 0,1 н. раствора NaOH, пошедший на титрование стандартной пробы, см3; k — коэффициент пересчета на точно 0,1 н. раствор NaOH; 1,4 — количество азота, соответствующее 1 см3 точно 0,1 н. раствора NaOH, мг; т — масса навески исследуемого вещества (продукта), г.
Расхождение между параллельными определениями не должно превышать 0,5 мг%.
Определение содержания гликогена в мясе рыбы и нерыбных объектах промысла. Гликоген — животный крахмал (С6Н10О5)N — полисахарид разветвленной структуры. Средний молекулярный вес 105...107. Состоит из остатков глюкозы в форме o-D-глюкопиранозы. Гликоген содержится в органах животных, в том числе рыб, и представляет собой резервное вещество. Легко расщепляется с образованием глюкозы, а при гидролизе с образованием молочной кислоты. Наиболее богаты гликогеном печень (до 20% на сырую массу) и мышцы (около 4% на сырую массу), очень богато им мясо беспозвоночных и моллюсков, например, в мясе мидий и устриц его содержится от 6 до 30% (на сухое вещество).
Метод определения содержания гликогена основан на его выделении из материала путем обработки последнего 30%-ным раствором щелочи с последующим гидролизом раствором НС1 для перевода в глюкозу.
Навеску исследуемою материала массой 2...4 г, взвешенную с погрешностью не более 0,0001 г, следует поместить в центрифужную пробирку, в которую предварительно налить 4...8 см3 30%-го раствора КОН. Пробирку неплотно прикрыть стеклянной пробкой и поместить (для гидролиза материала) в кипящую водяную баню на 3 ч. Через каждые 5...10 мин пробирку встряхивать. По окончании гидролиза (масса стала однородной) в пробирку добавить (при перемешивании ее содержимого стеклянной палочкой) 10 см3 90%-го спирта и снова поместить ее в водяную баню. Когда содержимое пробирки начнет кипеть, нагревание прекратить. После охлаждения уплотнить выпавший осадок гликогена центрифугированием и слить жидкость, образовавшуюся над осадком. При выпадении окрашенного осадка подвергнуть его вторичной обработке 30%-ным раствором КОН (при нагревании) и осаждению спиртом, как описано выше. Выделенный осадок гликогена промыть непосредственно в центрифужной пробирке сначала 96%-ным спиртом, а затем эфиром. После центрифугирования осторожно слить с осадка спирт и эфир и на небольшое время поместить пробирку на водяную баню для испарения остатка растворителей.
К осадку гликогена в пробирке следует добавить 6 см3 горячей дистиллированной воды, а затем нейтрализовать смесь по лакмусу, добавляя к ней сначала 2...3 капли концентрированной НС1, а затем 2,2%-ный ее раствор. После нейтрализации в пробирку внести 20 см3 2,2%-го раствора НС1, прикрыть ее стеклянной пробкой и поместить на 3 ч в кипящую водяную баню для гидролиза гликогена (превращения его в глюкозу). По окончании нагревания содержимое пробирки количественно перенести, смывая дистиллированной водой, в мерную колбу на 50 см3, нейтрализовать по лакмусу раствором КОН и довести объем содержимого, добавляя дистиллированную воду, до метки. После тщательного перемешивания содержимое колбы отфильтровать. 5 см3 фильтрата внести в обычную пробирку размером 25 х 200 мм и добавить 5 см3 окислительного реагента (см. ниже), смывая им со стенок пробирки капли исследуемого раствора. Если исследуемый раствор содержит очень большое количество гликогена, взять меньше фильтрата (2...3 см3), но обязательно прибавить к нему такое количество дистиллированной воды, чтобы объем исследуемой жидкости в пробирке составлял 5 см3. Хорошо перемешав содержимое пробирки, поместить ее на 20 мин в сильно кипящую баню, а затем быстро охладить водопроводной водой под краном. В охлажденную пробирку осторожно (без перемешивания) по стенке внести 1 см3 2,5%-го раствора KJ, а затем быстро добавить 3 см3 1 н. раствора H2SO2 при энергичном перемешивании смеси (встряхивание пробирки) и закрыть пробирку пробкой. Через 3 мин оттитровать выделившийся йод 0,01 н. раствором тиосульфата натрия (гипосульфита) в присутствии крахмала. Параллельно провести контрольный опыт. Содержание гликогена Х (в %) вычисляется по формуле:
x = (v-v1)*k*0.25*50*100 / m*v2*1000
где v — объем 0,01 н. раствора тиосульфата натрия, пошедший на титрование в контрольном опыте, см3; v, — объем 0,01 н. раствора тиосульфата натрия, пошедший на титрование в рабочем опыте, см3; v2 — объем фильтрата, взятый для обработки окислительным реагентом, см3; k — коэффициент пересчета на точнo 0,01 н. раствор тиосульфата натрия; 0,25 — количество (С6Н10О5)N, эквивалентное1 см3 0,01 н. раствора тиосульфата натрия, мг; 50 — объем всей жидкости в мерной колбе, полученный после гидролиза осадка (С6Н10O5), см3; т — масса навески исследуемого материала, г; 1000 — пересчет миллиграммов в граммы.
Расхождение между параллельными определениями не должно превышать 0,5%.
Примечание. Для проведения опыта должен быть приготовлен окислительный реагент — 28 г двузамещенного фосфата натрия (Na2HP04) и 40 г сегнетовой соли (калиево-натриевая соль винной кислоты — KNaC4H406 • 4Н20); их следует растворить в 500 см3 дистиллированной воды. К полученному раствору добавить 100 см3 1 н. раствора NaOH, прилить при помешивании 80 см3 10%-го раствора сернокислой меди (CuSO4 • 5Н20) и добавить 180 г сульфата натрия (Na2SO4). После растворения Na2SO4 жидкость перенести в мерную колбу на 1000 см3, добавить 50 см3 0,1 н. раствора KI и довести объем жидкости до метки, добавляя дистиллированную воду. Полученный раствор отстаивать в течение одного-двух дней, отфильтровать и хранить в плотно закрытой склянке из темного стекла. Реактив пригоден к употреблению при работе с растворами глюкозы концентрации не более 0,5 мг в 5 см3.
Похожие работы
... микроскопом: определяли наличие паразитов и их вид. При исследовании рыб общепринятым методом их не удавалось обычно сохранить живыми, тогда как после проведения исследования прижизненным методом рыбы во всех случаях оставались неповрежденными. Лишь в отдельных случаях при использовании жестких кисточек с искусственным волосом наблюдались небольшие кровотечения из жабр, которые быстро ...

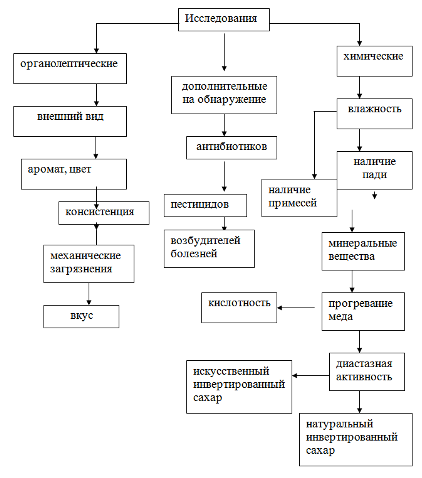
... при температуре 40—45 °С. В процессе фильтрации кусочек соты несколько раз переворачивают для более полного стекания воды. Каждую пробу исследуют отдельно. 3. Органолептический метод исследования Органолептическое исследование — оценка качества продукции с помощью органов чувств: обоняния, вкуса, осязания, зрения. Это исследование позволяет произвести предварительную оценку данного продукта. ...
... фитопланктона, называют антропогенным эвтрофированием водоемов. Подчеркивая всю важность биоиндикационных методов исследования, необходимо отметить, что биоиндикация предусматривает выявление уже состоявшегося или происходящего загрязнения окружающей среды по функциональным характеристикам особей и экологическим характеристикам сообществ организмов. Постепенные же изменения видового состава ...
... представление о величине глазных яблок (буфтальме, микрофтальме), размерах роговицы (микро и макрокорнеа). Глубине передней камеры, размерах и реакции на свет зрачка, состоянии области зрачки (мидриазе, колобоме) и пр. Методы исследования век, соединительной оболочки, роговицы, радужной оболочки. Начинают исследование больного с осмотра век, при котором устанавливают состояние кожи и краев век, ...
0 комментариев