Роль рецепторов плазматической мембраны в процессах агрегации тромбоцитов человека
Биологическая активность производных адамантана
Влияние производных адамантана на серотонин-индуцируемую агрегацию тромбоцитов человека in vitro
Механизмы влияния производных адамантана на индуцируемую агрегацию тромбоцитов человека in vitro
Stanford S. C. 5-Hydroxytryptamine. // Neurotransmitters, Drugs and Brain Function. – 2001. – 57, N 45. Р. 187-209
Навигация
Влияние производных адамантана с различными характерами заместителей на индуцированную агрегацию тромбоцитов человека
Влияние производных адамантана с различными характерами заместителей на индуцированную агрегацию тромбоцитов человека
83652
знака
29
таблиц
15
изображений
Введение
В медицине всё ещё стоит вопрос об эффективности лечения патологий системы свёртывания крови, в частности таких нарушений, как тромбоцитоз (патологическое увеличение числа тромбоцитов) и тромбоцитопения (уменьшение числа тромбоцитов в крови). Повышенная способность тромбоцитов к агрегации наблюдается при ряде заболеваний: ишемической болезни сердца, атеросклерозе, диабетических ангиопатиях и др., что значительно осложняет течение болезни [6, 43]. Путём направленного фармакологического воздействия можно снижать патологически возникшую гиперагрегацию тромбоцитов, или наоборот, повышать их способность к агрегации.
В данной работе исследовано влияние производных адамантана с различными характерами заместителей на индуцированную агрегацию тромбоцитов человека. Ранее было показано, что амиды N-[(адамантоил-1)-фенил]-антраниловой кислоты проявляли выраженное ингибирование серотонинового отёка, предполагалось, что это явление вызвано блокированием серотониновых рецепторов сосудистой стенки этими амидами [11].
Научная новизна работы: впервые исследованы вновь синтезированные производные адамантана, способные усиливать или снижать эффект известных индукторов агрегации тромбоцитов посредством модуляции функционирования соответствующих тромбоцитарных рецепторов.
Результаты работы имеют практическое значение – исследованные соединения после более детального изучения механизма их действия могут быть использованы в качестве медицинских препаратов.
По теме дипломной работы опубликовано три статьи (две из них приняты в печать). Результаты представлялись на 56 и 57 научных студенческих конференциях.
Цель работы: изучить механизмы влияния вновь синтезированных производных адамантана на индуцированную агрегацию тромбоцитов человека.
Задачи исследования:
1. Выявить среди исследуемых соединений синергистов (или агонистов) и антагонистов серотонина;
2. Выяснить механизмы влияния этих соединений на индуцируемую агрегацию тромбоцитов человека;
3. Проверить наличие свойств агонистов агрегирующих тромбоциты веществ у всех исследуемых соединений;
4. Выявить синергистов и антагонистов серотонина;
5. Выявить синергистов и антагонистов АДФ;
6. Выявить синергистов и антагонистов адреналина.
Глава 1. Обзор литературы
1.1. Механизмы агрегации тромбоцитов человека
Тромбоциты – плоские безъядерные клетки крови неправильной округлой формы, образующиеся из мегакариоцитов костного мозга путём отщепления от них участков цитоплазмы. Образование тромбоцитов регулируется гликопротеиновым гормоном тромбопоэтином. Нормальные тромбоциты – клетки диаметром 1-5 мкм, толщиной 0,5-0,75 мкм. Содержание тромбоцитов в крови здорового человека составляет 180-320 тыс. в 1 мл. Молодые тромбоциты проходят период созревания – восемь суток, зрелые циркулируют в кровотоке от пяти до 11 суток, с последующим разрушением в печени и селезёнке. Число тромбоцитов в крови относительно постоянно, но ночью оно снижается, а возрастает при пищеварении, тяжёлой мышечной работе, беременности [31].
На мембране тромбоцитов адсорбируются факторы свёртывающей системы крови. Тромбоциты способны выделять из фосфолипидов своих клеточных мембран арахидоновую кислоту и превращать её в тромбоксаны (при участии тромбоксансинтетазы), которые повышают агрегационную активность тромбоцитов. Примыкающая к оболочке тромбоцита область неструктурированной цитоплазмы – гиаломер. В гиаломере после активации тромбоцита при электронной микроскопии становятся видны микрофиламенты из актина, миозина и тропомиозина, составляющие вместе с микротрубочками цитоскелет клетки, который определяет её форму и способность перемещать органеллы, прикрепляться к поверхностям и образовывать псевдоподии [42, 48]. Актиновые филаменты имеют диаметр 3-5 нм, а миозиновые 6-10 нм. Микрофиламенты контактируют между собой, плазматической мембраной и мембранами гранул [85]. Микротрубочки в основном состоят из тубулина, их диаметр 25-30 нм. Концентрическая система из 5-30 микротрубочек тромбоцитов образует периферический пучок под плазматической мембраной вдоль экватора клетки. Также присутствуют и другие немембранные структуры – гранулы гликогена.
В тромбоцитах различают две системы мембран: плотную тубулярную систему (ПТС) и систему открытых каналов. ПТС – система узких трубочек диаметром 50 нм, подобная ЭПР скелетной мышцы. Эти трубочки не контактируют с системой открытых каналов, органеллами и плазматической мембраной, расположены и по экватору клетки и среди органелл [36, 86]. Система открытых каналов сообщается с гранулами тромбоцитов и с плазматической мембраной, за счёт чего и осуществляется секреция тромбоцитами специфических соединений в процессе реакции высвобождения. Поверхность тромбоцитов может увеличиваться за счёт системы открытых каналов [36, 82, 86]. Центральная часть тромбоцитов (грануломер) содержит органеллы и множество гранул различной структуры, формы и величины («электроноплотные» гранулы, α-гранулы, лизосомы). В электроноплотных гранулах содержатся АДФ, АТФ, ГТФ, ГДФ, неорганический фосфат, серотонин, Са2+; в α-гранулах – трансформирующий ростовой фактор, β-тромбоглобулин, антигепарин (фактор пластинок 4), фибронектин, альбумин, фибриноген, проакцелерин (фактор V), антиген фактора Виллебранда (фактор VIII), калликреин, α2-антиплазмин, тромбоспондин, гистамин; в лизосомах – кислые гидролазы (β-гексозаминидаза, β-галактозидаза, β-глюкуронидаза, β-арабинозидаза, β-глицерофосфатаза, арилсульфотаза) [22, 42]. В тромбоцитах имеются гликолитические ферменты и ферменты пентозофосфатного цикла, цикла лимонной кислоты и дыхательной цепи, АТФаза [42].
Важнейшим свойством тромбоцитов является способность к агрегации, в процессе которой в них синтезируется, а затем секретируется ряд белков и биологически активных веществ, стимулирующих свёртывание крови (гемостаз). Тромбоциты принимают участие и в защите организма от чужеродных агентов. Они обладают фагоцитарной активностью, содержат IgG, являются источником лизоцима и β-лизинов, способных разрушать мембрану некоторых бактерий.
Гемостаз осуществляется в 4 фазы: сокращение сосуда, образование тромбоцитарной пробки, формирование красного тромба и его ретракция, полное или частичное растворение тромба [29]. Различают внешний путь свёртывания (в месте повреждения сосудов, индуцируется коллагеном) и внутренний путь свёртывания (в области замедленного кровотока на патологически изменённой поверхности сосудистой стенки, индуцируется факторами плазмы), они различаются начальными этапами, объединяясь на стадии активации фактора Х. В настоящее время известно много факторов свёртывания крови, отсутствие любого из которых может привести к нарушению процесса коагуляции. В основном они представляют собой протеолитические ферменты (факторы XII, XI, X, IX, VII, II и калликреин – сериновые протеазы), присутствующие в крови в неактивной форме в виде проферментов. В процессе свёртывания они активируют друг друга в каскадной последовательности реакций. Активированные факторы обозначаются прибавлением буквы «а», например, Xа. (рис. 1)
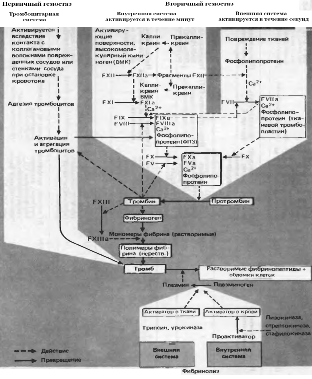
Рис. 1. Схема свёртывания крови и фибринолиза. ФП 3 – тромбоцитарный фактор 3 (по Циммерману, 1996)
Сразу после травмы наблюдается первичный спазм кровеносных сосудов, благодаря чему кровотечение в первые секунды может не возникнуть или носит ограниченный характер. Первичный спазм сосудов обусловлен выбросом в кровь в ответ на болевое раздражение адреналина и норадреналина и длится 10-15 с. Повреждение сосудов сопровождается немедленной активацией тромбоцитов вследствие появления высоких концентраций АДФ (из травмированных клеток сосудов), а так же обнажением субэндотелия, коллагеновых и фибриллярных структур [57]. В дальнейшем наступает вторичный спазм, обусловленный активацией тромбоцитов и выбросом в кровь сосудосуживающих агентов – серотонина, тромбоксана А2, адреналина и др. (реакция высвобождения).
Просвет повреждённых сосудов уменьшается и перекрывается массой тромбоцитов, прилипших к коллагеновым волокнам. Тромбоксан А2 и АДФ, выделяющиеся при разрушении внутриклеточной структуры активированных тромбоцитов, вызывают присоединение новых тромбоцитов. АДФ также может высвобождаться из разрушенных эритроцитов [32], и способствует превращению кровяных пластинок в диски, а также вызывает секрецию тромбоцитарных белков из электроноплотных и α-гранул [68]. Тромбоциты распластываются, у них появляется до 10 «отростков», которые могут в 5-10 раз превышать диаметр самой клетки [2, 34]. Организованное кольцо микротрубочек распадается, изменяется распределение микрофиламентов и микротрубочек, микротрубочки могут образовывать клубочки [36, 54].
На нескольких из этапов гемокоагуляции существуют перекрёстные взаимодействия между внешней и внутренней системами свёртывания, благодаря чему обеспечиваются «альтернативные» пути для процессов коагуляции [42, 65].
Агрегация тромбоцитов наступает одновременно с их адгезией, это активный процесс, протекающий с затратой энергии. Адгезия тромбоцитов к волокнам соединительной ткани по краям раны обусловлена олигомерным гликопротеином, содержащимся в субэндотелии и кровяных пластинках, – фактором Виллебранда. Он образует мостики между субэндотелиальными структурами и специфическими рецепторами (гликопротеин Ib) на мембране тромбоцитов [8]. Фибриноген – белок, содержащийся в плазме и тромбоцитах, тоже способный образовывать между тромбоцитами связующие мостики, что приводит к появлению сети фибрина и тромбоцитарной пробки. Частичный протеолиз молекулы фибриногена наступает под действием тромбина. Для образования ковалентных связей необходим фибринстабилизирующий фактор XIII (трансглутаминаза), активируемая тромбином в присутствии ионов кальция. Трёхмерная сеть волокон фибрина удерживает клетки крови, формируется красный тромб (рис. 1). Благодаря контрактильному белку тромбостенину тромбоциты подтягиваются друг к другу, тромбоцитарная пробка сокращается и уплотняется, т. е. наступает её ретракция [49]. Сгусток становится более плотным и стягивает края раны, что облегчает её зарастание клетками соединительной ткани. В норме остановка кровотечения из мелких сосудов занимает от двух до четырёх минут [34].
Таким образом, при повреждении кровеносного сосуда в крови появляются вещества, инициирующие процесс агрегации тромбоцитов. Характер действия этих веществ разный, так, слабыми агрегирующими агентами являются АДФ, серотонин, адреналин, вазопрессин, – они действуют первыми, лишь запуская каскад последующих реакций. Более быстро агрегация происходит под действием коллагена, тромбина, ионофора А23187. Агрегация тромбоцитов может носить обратимый характер (вслед за агрегацией наступает дезагрегация, распад агрегатов), что зависит от недостаточной дозы агрегирующего агента или от наличия в крови простациклина PGI2, синтезирующегося в эндотелии сосудов, который ингибирует агрегацию тромбоцитов [5]. Травма сосуда всегда сопровождается образованием сразу нескольких индукторов агрегации, способных инициировать агрегацию тромбоцитов разными путями, и их концентрация достаточно высока, поэтому дезагрегация, как правило, не случается.
Биохимические механизмы активации тромбоцитов с их последующей агрегацией различны, что обусловлено различным характером инициаторов агрегации. Однако, все известные агреганты так или иначе способствуют увеличению концентрации Са2+ в цитоплазме тромбоцита.
Общее содержание Са2+ в тромбоцитах составляет около 60 нмоль/109 клеток. Четверть этого кальция связана с мембранами тромбоцитов, но значительное его количество содержится в тубулярной системе и плотных гранулах. Концентрация свободных ионов Са2+ в цитоплазме тромбоцитов (10-7 моль/л)почти в 1000 раз ниже, чем в плазме (10-3 моль/л). Такая разница в концентрации этих ионов по обе стороны плазматической мембраны поддерживается мембранными Са2+-АТФазами, которые используют энергию гидролиза АТФ для откачивания Са2+ из цитоплазмы в окружённые мембранами клеточные органеллы, а так же аденилатциклазами и фосфодиэстеразами, контролирующими уровень цАМФ. Активированная аденилатциклаза катализирует образование цАМФ из АТФ, фосфодиэстераза осуществляет гидролиз цАМФ до 5-АМФ. Аденилатциклаза тромбоцитов локализована на внутренней стороне плазматической мембраны, на мембранах плотной тубулярной системы и системы открытых каналов. Фосфодиэстераза находится в цитоплазме. Активация аденилатциклазы происходит посредством взаимодействия простагландинов Е1, I2 и D2 со специфическими рецепторами мембран, сопряжёнными с ГТФ-связывающими белками (G-белки). Для этих рецепторов цАМФ служит внутриклеточным медиатором. Активированные рецепторы действуют на G-белки, которые стимулируют аденилатциклазу, образуется цАМФ, активируются цАМФ-зависимые ферменты (протеинкиназы), катализирующие фосфорилирование небольшой группы тромбоцитарных белков [36].
Адреналин, связываясь со специфическими рецепторами мембраны тромбоцитов, посредством G-белков стимулирует образование тромбоксана А2 [36]. Предполагается, что ускоряющий агрегацию тромбоцитов эффект адреналина связан с модуляцией мембран при его взаимодействии с α-адренорецепторами и изменением её проницаемости к ионам Са2+ [73, 79]. Показано, что антагонисты α2-адренорецепторов способны блокировать агрегацию, а агонисты усиливают её, поскольку активированные Gi-белки ингибируют аденилатциклазу [10, 59]. Через β-адренорецепторы происходит замедление агрегации тромбоцитов. Стимуляция β-адренорецепторов посредством Gs-белков приводит к активации аденилатциклазы, т. е. катехоламины способны регулировать активность тромбоцитов, влияя на уровень цАМФ в клетке. Адреналин вызывает агрегацию тромбоцитов без изменения их формы [53]. Адреналин-индуцированное изменение оптической плотности проб наблюдается через 30 с после его добавления [36].
Активация тромбоцитов тромбином, коллагеном, фактором активации тромбоцитов (ФАТ) и некоторыми другими агрегантами происходит через образование из фосфолипидов плазматической мембраны (фосфатидилхолина и фосфатидилинозитола) арахидоновой кислоты [50]. Активированная через сопряжённые с Gs-белками рецепторы для ФАТ фосфолипаза С катализирует этот процесс. Арахидоновая кислота способна метаболизировать, вновь включаться в фосфолипиды или диффундировать из тромбоцитов. Высвобождение арахидоновой кислоты из фосфолипидов тромбоцитов могут катализировать также фосфолипаза А2 и глицеролипаза. Образовавшаяся арахидоновая кислота может окисляться двумя ферментами: цитоплазматической циклооксигеназой (ЦОГ) и мембранносвязанной липоксигеназой (ЛОГ). При действии ЦОГ образуются нестабильные циклические эндопероксиды, которые превращаются в простагландины G2 и Н2, тромбоксан А2, простациклин с дальнейшим образованием из них 6-кето-простагландина Е1, 12-гидрокси-5,8,10-гептадекатриеновой кислоты, тромбоксана В2 и малондиальдегида. Конечными продуктами липоксигеназного пути окисления арахидоновой кислоты являются 12-гидрокси-5,8,10,14-эйкозатетраеновая кислота (12-НЕТЕ) и лейкотриены [36]. Образующиеся из арахидоновой кислоты простагландины и тромбоксан А2 (ионофор кальция) являются агрегантами. Простациклин, синтезируемый клетками эндотелия кровеносных сосудов, напротив, мощный ингибитор агрегации тромбоцитов.
Связывание АДФ со специфическими рецепторами плазматической мембраны тромбоцита приводит к увеличению концентрации ионов кальция в цитоплазме клеток (увеличивается его поступление из внешней среды и из внутриклеточных запасов) [33]. Кроме того, происходит активация фосфолипазы А2, которая стимулирует высвобождение арахидоновой кислоты из фосфолипидов мембраны и образование из неё тромбоксана А2 – активного проагреганта и вазоконстриктора. Форма тромбоцитов изменяется без сопутствующей активации фосфолипазы С, что сопровождается ингибированием аденилатциклазы [61, 77]. Реализация метаболических эффектов активации АДФ-рецепторов тромбоцитов осуществляется через сопряжение с мембранным G-белком. Сегодня известно, что при активации на поверхности каждого тромбоцита появляется до 50–100 тысяч гликопротеиновых рецепторов [76]. Экспериментальные данные многих клинических исследований свидетельствуют, что удаление из среды АДФ или связывание АДФ-рецепторов с конкурентным антагонистом существенно угнетает агрегацию тромбоцитов [72, 76, 78, 81].
Для серотонина на поверхности тромбоцитов существует два типа рецепторов – ионные каналы и рецепторы, сопряжённые с G-белком [40, 45]. Согласно [47], механизм действия серотонина состоит в регуляции цАМФ-зависимых путей, и поэтому реакция на серотонин способна блокироваться стимуляторами аденилатциклазы и ингибиторами фосфодиэстеразы. Серотонин стимулирует поступление ионов кальция из плазмы в тромбоциты через рецепторуправляемые каналы для двухвалентных катионов, запуская фосфатидилинозитоловый цикл с образованием фосфатидной кислоты, являющейся кальциевым ионофором. Активация G-белков, сопряжённых с другим типом серотониновых рецепторов приводит к активации фосфолипазы С, которая гидролизует трифосфатидилинозитол (или фосфатидил-4,5-бифосфат) в дифосфатидилинозитол (фосфатидилинозитол-4-фосфат) с образованием 1,2-диацилглицерола (ДАГ), который фосфорилируется в фосфатидную кислоту (ФК). Это самые ранние этапы активации тромбоцитов. Диацилглицерол стимулирует Са2+-активируемые фосфолипидзависимые протеинкиназы (в частности, ПК С), которые фосфорилируют белки. Фосфатидная кислота – кальциевый ионофор, высвобождает кальций из внутриклеточных депо, присоединяя к своим фосфатным группам. Ионы кальция стимулируют работу актина и миозина микрофиламентов, в результате чего гранулы тромбоцитов перемещаются к системе открытых каналов, осуществляется реакция высвобождения собственных агрегантов, что усиливает собственный ответ клетки и активирует другие тромбоциты.
Таким образом, активация тромбоцитов начинается при взаимодействии агрегантов с рецепторами плазматической мембраны. Важную роль в передаче сигнала внутрь тромбоцита играют G-белки плазматической мембраны, а также изменение проницаемости мембраны для ионов (если рецептор является ионофором) [38].
0 комментариев