Навигация
Заболевание Фенилкетонурия (ФКУ)
28011
знаков
7
таблиц
0
изображений
Заболевание Фенилкетонурия (ФКУ)
Актуальность
1. Фенилкетонурия (ФКУ) является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10000, однако значительно варьирует в зависимости от популяции: от 1:4560 в Ирландии, до 1:100.000 в Японии. (см. таблицу)
Табл. Заболеваемость ФКУ по данным массового скрининга
| Москва | 1:11765 |
| Белоруссия | 1:5578 |
| Хабаровский край | 1:9708 |
| Англия | 1:14306 |
| ФРГ | 1:6697 |
| Ирландия | 1:4560 |
| Греция | 1:18460 |
| Шотландия | 1:8350 |
| США | 1:15059 |
| Австралия | 1:11224 |
| Мексика | 1:45610 |
| Япония | 1:100000 |
| ЧСФР | 1:8122 |
| Польша | 1:9248 |
2. Ребенок с фенилкетонурией выглядит при рождении здоровым. Отставание психического развития может происходить постепенно и стать очевидным лишь через несколько месяцев. Установлено, что нелеченный ребенок теряет около 50 баллов IQ к концу первого года жизни. Отставание психического развития обычно довольно выражено, и большинство детей нуждаются в социальной помощи.
3. В связи с тяжестью клинических проявлений и возможностью профилактического лечения ФКУ рекомендована для выявления среди новорожденных. Ранняя диагностика позволяет своевременно начать лечение и предотвратить инвалидизацию больного.
4. Лечение и реабилитация больных ФКУ требует значительных финансово-экономических затрат. Это обусловлено высокой себестоимостью продуктов лечебного питания, а также усилиями по социальной реабилитации больного.
5. Помимо этого, об актуальности заболевания, свидетельствует проблема материнской ФКУ, лишь недавно появившаяся в России. Наблюдается высокая частота умственной отсталости среди потомства женщин, страдающих ФКУ и не получающих диету в зрелом возрасте.
6. Скудная клиническая картина в раннем возрасте, постепенное развитие патологических изменений, приводит к серьезным затруднениям в ранней диагностике этого заболевания. В тоже время, диетотерапия больных с ФКУ должна находиться под тщательным динамическим контролем. А психоневрологическая реабилитация больных (медикаментозная, физиолечение, медико-педагогическая) требует индивидуального подхода. Педиатры, в чьи обязанности входит реабилитация больных ФКУ, встречаются с ней в повседневной практике нечасто. Поэтому необходимо ознакомить врачей с особенностями течения и лечением ФКУ.
Этиология, патогенез
По Mc Kusick выделяется несколько типов фенилкетонурии.
Фенилкетонурия I
Классическая фенилкетонурия. Была описана А. Folling в 1934 г. Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена ФАГ, локализующегося в длинном плече 12 хромосомы. В гене ФКУ выявлено 12 различных гаплотипов. При этом 90% всех генов ФКУ ассоциировано с четырьмя гаплотипами, из них гаплотип 3 характеризует около 38% всех генов ФАГ, а гаплотип 2 - около 20%.
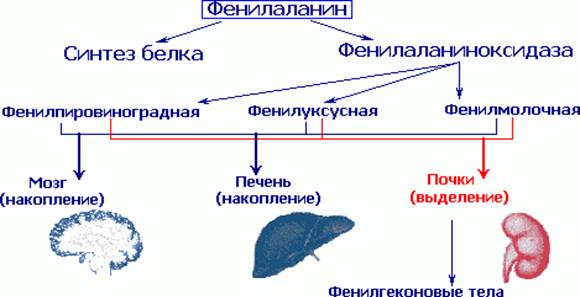
В основе болезни лежит дефицит фермента фенилаланин - 4 - гидроксилазы (ФАГ), обеспечивающей превращение фенилаланина в тирозин. В результате метаболического блока происходит значительное накопление в тканях и жидкостях больного организма фенилаланина и таких его производных, как фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин и др.
По мнению различных авторов в патогенезе ФКУ имеют значение следующие обстоятельства: прямое токсическое действие на центральную нервную систему фенилаланина и его производных, нарушения в обмене белков, липо- и гликопротеидов, расстройства транспорта аминокислот, нарушение метаболизма гормонов и др., а также перинатальные факторы. В последнее время все большее значение в патогенезе ФКУ придается нарушениям обмена моноаминовых нейромедиаторов (катехаламинов и серотонина). Известна исключительно важная роль этих медиаторов в функционировании центральной нервной системы. Исследования показали, что у больных резко снижено содержание конечных продуктов их метаболизма (гомованилиновой кислоты и 5-оксииндолуксусной кислоты) в крови, моче и цереброспинальной жидкости.
Определенное значение в генезе церебральных расстройств могут играть нарушения функции печени. У большинства больных ФКУ при обследовании обнаруживаются различные биохимические и морфологические изменения, свидетельствующие о заинтерсованности этого органа в патологическом процессе: диспротеинемия, генерализованная гипераминацидемия, повышение показателя дифениламиновой реакции, компенсированный метаболический ацидоз, признаки белковой и жировой дистрофии печени с нарушением окислительной и белоксинтезирующей функции клеточных органелл.
Фенилкетонурия II
Впервые атипичная ФКУ была описана I. Smith в 1974 г. S. Kaufman et al. В 1975 г. обнаружили дефицит дигидроптеридинредуктазы при этом состоянии.
Заболевание наследуется аутосомно-рецессивно. Генный дефект локализуется в коротком плече 4 хромосомы, участке 4р15.3. В результате недостаточности дигидроптеридинредуктазы нарушается восстановление активной формы тетрагидробиоптерина, участвующего в качестве кофактора в гидроксилировании фенилаланина, тирозина и триптофана. Вследствие этого развиваются метаболические блоки на путях превращения фенилаланина в тирозин, а также образования предшественников нейромедиаторов катехоламинового и серотонинового ряда L-дофы и 5-окситриптофана, что подтверждается резким снижением содержания в тканях и жидкостях больного организма (в том числе в мозге и цереброспинальной жидкости) их конечных продуктов - гомованилиновой и 5-оксиндолуксусной кислот.
Существенным для патогенеза заболевания является снижение уровня фолатов в сыворотке крови, эритроцитах и цереброспинальной жидкости. Это объясняется тесной взаимосвязью обмена фолатов и биоптерина, в частности участием дигидроптеринредуктазы в метаболизме тетрагидрофолиевой кислоты.
Фенилкетонурия III
Этот вариант болезни впервые описан S. Kaufman et al. в 1978 г. Заболевание наследуется аутосомно-рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин синтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин трифосфата. Ключевую роль в патогенезе играет дефицит тетрагидробиоптерина. Развивающиеся при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ II.
Другие варианты ФКУ
В последние годы стали известны другие формы атипичной ФКУ, связанные с дефицитом тетрагидробиоптерина.
Недостаточность гуанозин 5-трифосфат циклогидролазы (S. Kaufman et al., 1987) описана по крайней мере у пяти больных. Этот фермент катализирует первую ступень синтеза тетрагидробиоптерина и при его дефиците в моче обнаруживается крайне низкая концентрация всех птеринов.
N. Blau и соавторы (1989) сообщили о новом варианте атипичной ФКУ - примаптеринурии - у двух детей с легкой гиперфенилаланинемией. Энзиматический дефект пока не известен. В моче обнаруживается в больших количествах продукт изомеризации биоптерина-7 - изобиоптерин (примаптерин) и некоторые другие его производные. Соотношение неоптерин/биоптерин у больных значительно повышено. Нагрузка тетрагидробиоптерином нормализует концентрацию фенилаланина в сыворотке и уровень неоптерина в моче, резко повышает экскрецию биоптерина и примаптерина. Отличием от других атипичных форм ФКУ является нормальная концентрация в цереброспинальной жидкости нейромедиаторных метаболитов - гомованилиновой и 5-оксииндолуксусной кислот.
Материнская ФКУ
В процессе изучения ФКУ было обращено внимание на высокую частоту умственной отсталости среди потомства женщин, страдающих ФКУ и не получающих диету в зрелом возрасте. Это состояние получило наименование материнской ФКУ. Патогенез патологии мало изучен, однако предполагается, что он сходен с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с уровнем фенилаланина в плазме матери. Причем в связи с накоплением этой аминокислоты в плаценте, ее содержание в организме плода оказывается выше, чем у матери. Тем не менее прямое токсическое действие фенилаланина точно не подтверждено. Появление признаков патологии у потомства не зависит от наличия или отсутствия умственной отсталости у женщин и не связано с развитием у детей ФКУ. Есть данные, что тяжесть заболевания у больных ФКУ, родившихся от матерей, также страдающих этой болезнью, не отличается от степени поражения их сибсов, не унаследовавших ФКУ.
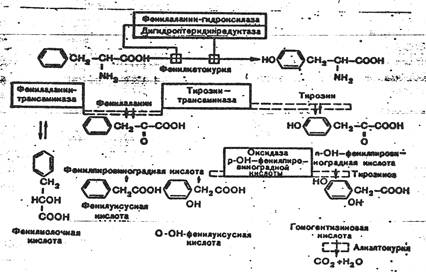
Схема 1. Пути обмена фенилаланина и его предшественников, и “точки приложения” метаболических дефектов.
гуанозин 5’ трифосфат
![]() моноаптерин
моноаптерин
дигидронеоптерин
![]()
![]() 1
1
дигидронеоптерин трифосфат
![]()
неоптерин
2
6-пировоилтетрагидроптерин
![]()
3
фенилаланин
тирозин тетрагидробиоптерин
![]()
![]() триптофан
триптофан
![]()
| 4 4 |
5 птерин
![]()
тирозин
![]() 5-окситриптофан
дигидробиоптерин 7,8-дигидробиоптерин
5-окситриптофан
дигидробиоптерин 7,8-дигидробиоптерин
![]() дофа
дофа
![]()
дофамин биоптерин
Пояснение к схеме 1.
1. гуанозин 5’ трифосфат циклогидролаза
2. 6-пирувоилтетрагидроптерин синтаза
3. сепиаптерин редуктаза
4. фенилаланин-4-гидроксилаза, тирозингидроксилаза, триптофангидроксилаза
5. дигидроптеридин редуктаза
Клиническая картина
Фенилкетонурия I
Манифестация ФКУ происходит на первом году жизни, обычно в возрасте 2-6 мес. Первыми проявлениями болезни служат вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), судороги, признаки аллергического дерматита. Появляется характерный “мышиный” запах. Отчетливо формируется задержка статикомоторного и психоречевого развития. При отсутствии лечения умственная отсталость достигает, как правило, глубокой степени.
Ранним симптомом заболевания может стать рвота, иногда настолько сильная, что ее ошибочно расценивают как проявление пилоростеноза. В более старшем возрасте нелеченные дети становятся гиперактивными, осуществляют бесцельные движения, ритмические покачивания, у них определяется атетоз.
При физикальном исследовании обращает на себя внимание то, что ребенок выглядит более белокурым, чем его здоровые сиблинги: у него светлая кожа и голубые глаза. У некоторых больных появляется себорейная или экзематоидная кожная сыпь, обычно умеренно выраженная и исчезающая по мере роста ребенка. От них исходит необычный запах фенилуксусной кислоты, который характеризуют как заплесневелый, мышиный или волчий. Не встречается характерных неврологических изменений, однако у большинства детей определяются гипертонус и повышение глубоких сухожильных рефлексов. Около 1/4 детей страдают судорогами и более чем у 50% появляются изменения на ЭЭГ. Часто у нелеченных детей определяют микроцефалию, выступающую верхнюю челюсть с широко расставленными зубами, гипоплазией эмали, отставание роста. Клинические проявления классической фенилкетонурии редко встречаются в странах, в которых эффективно действует программа неонатального скрининга на это заболевание.
Фенилкетонурия II
В клинической картине ФКУ II преобладает тяжелая умственная отсталость, судороги, признаки повышенной возбудимости, сухожильная гиперрефлексия, мышечная дистония, спастический тетрапарез. Течение болезни прогрессирующее и нередко приводит к смерти в 2-З-летнем возрасте. Больные, как правило, выявляются в результате массового скрининга новорожденных на ФКУ. Уровень фенилаланина в крови у этих детей увеличен, иногда может превышать 1200 мкмоль/л. Появление клинической симптоматики, как правило развивается в начале второго полугодия жизни, несмотря на диетотерапию.
Фенилкетонурия III
Клиническая картина болезни напоминает ФКУ II и включает тяжелую умственную отсталость, микроцефалию, спастический тетрапарез. Для подтверждения диагноза требуется:
· исследование биоптеринов мочи (повышенная экскреция дигидронеоптерин трифосфата и продукта его распада - неоптерина, низкий уровень биоптерина, резкое увеличение соотношения неопте рин/биоптерин).
· пероральный нагрузочный тест с тетрагидробиоптерином (падение уровня фенилаланина с одновременным повышением концентрации тирозина в крови);
· энзиматическое исследование (дефицит 6-пирувоилтетрагидроптерин синтазы в эритроцитах или гепатоцитах при сохранной активности фенилаланингидроксилазы и тетрагидробиоптеринредуктазы). Последнее исследование пригодно для выявления гетерозиготного носительства. Пренатальная диагностика ФКУ III теоретически возможна на основании определения активности ключевого фермента в эритроцитах плода или анализа птеринов и аминокислот в амниотической жидкости.
Возможности диагностики
Диагностика основывается на совокупности генеалогических данных (возможны эндогамный брак, аналогичная патология у сибсов), результатов клинического и биохимического обследования. Уровень фенилаланина в крови у больных превышает 900-1200 мкмоль/л. В моче присутствуют продукты трансаминирования и декарбоксилирования фенилаланина, в первую очередь, фенилпировиноградная кислота.
Проба Феллинга
Выявление фенилпирувата в моче при помощи хлорида 3-х валентного железа. Метод недостаточно точный и позволяет диагностировать заболевание только со 2-го месяца жизни.
Тест Гатри
Методику теста описал Роберт Гатри в 1961 году. В ее основе лежит бактериальное ингибирование определенных штаммов Bacillus cereus.
Хроматография
Проводится тонкослойная хроматография аминокислот плазмы крови и мочи.
Флуориметрия
С помощью современных автоматических флуориметров, возможно проведение массового автоматизированного скрининга.
Определение активности ФАГ или тетрагидробиоптеринредуктазы
Значительное снижение активности ферментов, обнаруживается у гомозигот, а умеренное (до 70%) у гетерозигот по мутантному аллелю ФКУ.
Поиск мутантного гена
Проводится прямой поиск мутантного гена с помощью синтетических олигонуклеотидных зондов. Помимо этого возможно прямое определение мутаций в гене фенилаланингидроксилазы или косвенное подтверждение наследования мутантного локуса ФАГ в одной или двух копиях, идентифицируемое по ПДРФ (полиморфизм длины рестрикционных фрагментов) или ВНТР (вариабельные по количеству тандемные повторы) в ДНК пробанда и его родителей.
Скрининг
Клинический полиморфизм отклонения от классической картины, возможность проведения эффективного лечения при своевременной диагностике, диктует необходимость проведения скрининга. Целью скрининга является раннее выявление заболевания и назначение диетического лечения больным до достижения ими 8-недельного возраста. Эти сроки начала лечения по общепризнанному мнению позволяют обеспечить полноценное развитие детей.
Скрининговые исследования проводятся в два этапа:
Первый этап предусматривает обнаружение отклонений от нормы в содержании фенилаланина в сыворотке крови новорожденного.
Обследования проводятся на 4-5-й день жизни ребенка. Кровь наносится на бланк из специальной впитывающей бумаги.
Обязательное условие для выполнения скрининга: диаметр кровяного пятна должен быть равен диаметру круга на фильтрованной бумаге с пропитыванием кровью обратной стороны. Заполненные бланки направляются в ближайшую специализированную лабораторию.
Второй этап - повторное тестирование из того же пятна крови в случаях получения результата анализа на первом этапе с уровнем фенилаланина выше допустимого.
При повторно положительных тестах информация направляется в районные медико-генетические центры с целью немедленного приглашения ребенка к врачу для окончательного уточнения диагноза ФКУ.
В Российской Федерации скрининг-тесты осуществляются в 45 медико-генетических лабораториях различных регионов, куда поступают анализы крови всех новорожденных из всех родильных домов.
Если диагноз ФКУ подтверждается, необходимо сразу начинать лечение, это позволяет предотвратить тяжелые последствия в развитии ребенка.
Лечение
Элиминационная диета
Главным способом лечения ФКУ является диетотерапия, ограничивающая поступление в организм пищевого белка и фенилаланина до минимальной возрастной потребности. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты (саго, хлеб, вермишель, крупка, приготовленные на крахмальной основе). Однако, в период интенсивного роста и развития ребенка поступление белка в организм должно быть достаточным. Дефицит его незамедлительно отразится на процессе формирования всех органов и систем. Поэтому нельзя полностью исключить из рациона новорожденного материнское молоко. Для коррекции питания детям даются белковые гидролизаты, лишенные фенилаланина, но содержащие все другие необходимые аминокислоты. Больные нуждаются в дополнительном введении витаминов, особенно, группы В, минеральных веществ и микроэлементов.
Сразу после подтверждения диагноза ФКУ необходимо начать незамедлительное, но постепенное (в течение З-х дней) исключение из питания больного высокобелковых продуктов питания, заменяя их малобелковыми.
Если ребенок находится в возрасте до 1 - 1,5 мес и получает только грудное молоко или его заменитель - его постепенно приучают к лечебной молочной смеси, не меняя режима кормления.
Специальные лечебные продукты питания для больных ФКУ
Эти продукты производятся на основе гидролизата казеина или смеси аминокислот: Лофеналак, Фенил-Фри, Нофелан, отечественный Тетрафен и другие. Для грудных детей используется Лофеналак.
Табл. 1. Состав продукта “Лофеналак” на 100 грамм сухой смеси.
| Энергия | 462 ккал |
| Белки | 15 г |
| Жиры | 18 г |
| Углеводы | 60 г |
| Фенилаланин | 16мг/100ккал |
Витамины, минеральные вещества и необходимые для роста аминокислоты добавлены в таком количестве, чтобы смесь по составу максимально приближалась к обычным молочным смесям для детского питания.
Однако содержание фенилаланина в Лофеналаке не обеспечивает суточную потребность в этой аминокислоте. Поэтому рекомендуется готовить молочную лечебную смесь, используя 2 компонента: Лофеналак и грудное молоко или его заменитель, растворяя их в кипяченой воде.
Для детей с ФКУ старте 1 года принципы лечебной диеты не отличаются от диеты детей грудного возраста. Рацион состоит из:
Похожие работы
... . Другие варианты ФКУ: Эти формы ФКУ связаны с нарушением альтернативных путей обмена фенилаланина. Формируется метилминдальная ацидурия и парагидроскифенилуксусная ацидурия. · Материнская фенилкетонурия. Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих диету в зрелом возрасте. Патогенез мало изучен, предполагается, что он сходен с патогенезом остальных форм ...
... Сфингомиелиназа Сфингомиелин, холестерин. Лейкоциты, материал биопсии органов, фибробласты кожи (инфантильная и ювенильная формы) Материал биопсии Гиперлипопротеидемии (ГЛП) Наследственные заболевания нарушений липидного обмена представлены большим количеством нозологических форм, которые обусловливают развитие атеросклероза и других форм сердечно-сосудистой патологии. Впервые ...
... их зрение снижено и не улучшается с возрастом. Альбинизм встречается с частотой 1 на 39.000, наследуется по аутосомно-рецессивному типу (см. приложение рис. 1). Ген локализован на длинном плече 11-й хромосомы. Наследственные заболевания, связанные с нарушением обмена углеводов Известно, что углеводы входят в состав ряда биологически-активных веществ — гормонов, ферментов, мукополисахаридов ...
... , вызванные динамическими му-тациями.-----------------------T-----------T-------T-----T------T------T----------------------¬ Болезнь, номер по ¦ Ген, лока-¦Триплет¦Норма¦Прему-¦Мута- ¦Литература ¦ МакКьюсику (MIM) ¦ лизация ¦ ¦ ¦тация ¦ция ¦ ¦ -----------------------+-----------+-------+-----+------+------+----------------------+ Синдром ломкой X-хро- ¦FMR1, FRAXA¦(CGG)n ...
0 комментариев