Навигация
КОМПЬЮТЕРНЫЙ АНАЛИЗ ГЕНЕТИЧЕСКИХ ТЕКСТОВ
37995
знаков
0
таблиц
6
изображений
4. КОМПЬЮТЕРНЫЙ АНАЛИЗ ГЕНЕТИЧЕСКИХ ТЕКСТОВ.
Выявление и анализ закодированных в последовательностях функциональных сигналов требует применения современных методов информатики - качественных баз данных с современными средствами управления, новейших методов распознавания образов, статистических исследований, применения специальных алгоритмов для преодоления возникающих вычислительных трудностей.
В настоящее время исследование функциональных свойств расшифрованных последовательностей нуклеиновых кислот - это новый раздел молекулярной биологии, граничащий с информатикой, с одной стороны, и молекулярной биофизикой - с другой. Можно с уверенностью сказать, что в настоящее время анализ последовательности биополимера позволяет извлечь лишь очень небольшую долю закодированной в ней информации. В конечном счете точное выявление функциональных особенностей в последовательностях нуклеиновых кислот будет возможно только после детального исследования соответствующих реакций, осуществляемых нуклеиновобелковыми комплексами.
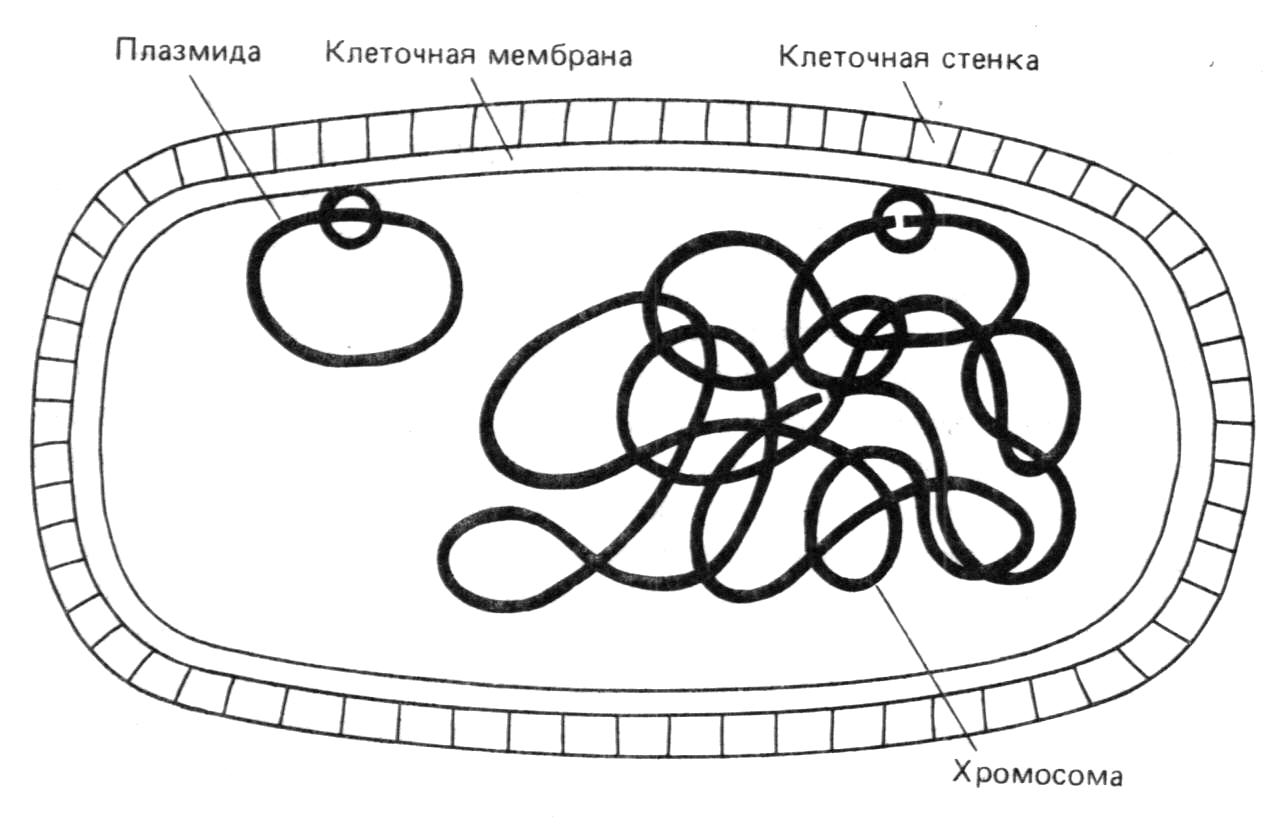
Для оперативной работы с последовательностями создаются специальные банки данных. В банке в доступном для пользователя виде хранится каждая расшифрованная последовательность и ее паспорт, в котором указаны различные сведения о ней. Это сведения об организме, из которого выделена последовательность, о документе, где она описана, о расположении на ней регуляторных участков и белках, которые она кодирует и т.д. В настоящее время созданы три большие базы данных последовательностей нуклеиновых кислот: "Genbank" (Лос-Аламос, США - более 30 млн. нуклеотидов), база данных нуклеотидных последовательностей Европейской молекулярно-биологической лаборатории (EMBL, Гейдельберг, ФРГ - более 30 млн. нуклеотидов) и "Генэкспресс" (СССР, ВИНИТИ-ИМГ АН СССР - более 11 млн. нуклеотидов). Известны также несколько белковых баз данных, наиболее представительной из которой является MBRF-PIR (США). Эти базы данных распространяются на различных носителях - магнитных лентах и дисках, на оптических дисках.
Кроме построения филогенетических древ геномов вирусов компьютерный анализ применяется при поиске гомологий, распознавании кодирующих областей, функциональных сигналов, физическом (рестрикционном) картировании молекул ДНК и для предсказания вторичных структур РНК.
Сейчас в мире создано большое количество программ ( обычно организованных в пакеты ) , предназначенных для анализа последовательностей нуклеиновых кислот и избавляющих исследователей от многих трудоёмких рутинных операций , в том числе: подсчёт числа моно -, ди – и тринуклеотидов, перевод нуклеотидной последовательности в аминокислотную и т.д.
Все программы условно делятся на два класса: общего назначения и специального. Первые осуществляют ряд_ наиболее распространенных операций по сбору и анализу последовательностей и позволяют: вводить и редактировать новые последовательности, считывать с помощью сканирующих устройств информацию непосредственно с автографов или гелей', находить участки узнавания эндонуклеаз рестрикции и представлять результаты в удобном (табличном или графическом) виде, находить участки с элементами поворотной и зеркальной симметрии (палиндромы), транслировать нуклеотидную последовательность в белковую во всех трех рамках считывания, сравнивать две последовательности методом точечных матриц гомологии, сравнивать новую последовательность со всеми данными Ген Банка, находить участки, обогащенные теми или иными нуклеотидами, вычислять гипотетическую температуру плавления ДНК, осуществлять автоматическую сборку секвенированных фрагментов в единую структуру - молекулу ДНК, транслировать белковую последовательность в нуклеотидную с учетом неравномерности использования кодонов-синонимов, определять молекулярную массу НК и белков, предсказывать вторичную структуру белков, вычислять свободную энергию образования шпилек и др.
Программы специального назначения создаются для решения более специальных и часто более сложных задач и представляют интерес для более узкого круга специалистов. Так, например, они могут выполнять ряд функций: вычисление длины фрагментов ДНК на основании их электрофоретической подвижности в гелях; выбор гибридизационных зондов; предсказание вторичной структуры РНК; локализация нуклеотидов в гене, которые могут быть изменены (без изменения аминокислотной последовательности) с целью введения сайта узнавания эндонуклеазы рестрикции; нахождение участков с потенциально возможной структурой Z-формы ДНК; выявление функционально значимых участков в неизвестной вновь расшифрованной структуре на основании ранее выведенного консенсуса (в результате сравнительного анализа ряда известных структур с одинаковой функцией); локализацию участков, кодирующих белки, и т.д.
Для примера представлено меню пакета программ MICROGENIE, из которых следует, какие функции общего или специального назначения может выбрать исследователь при работе с нуклеотидными последовательностями.
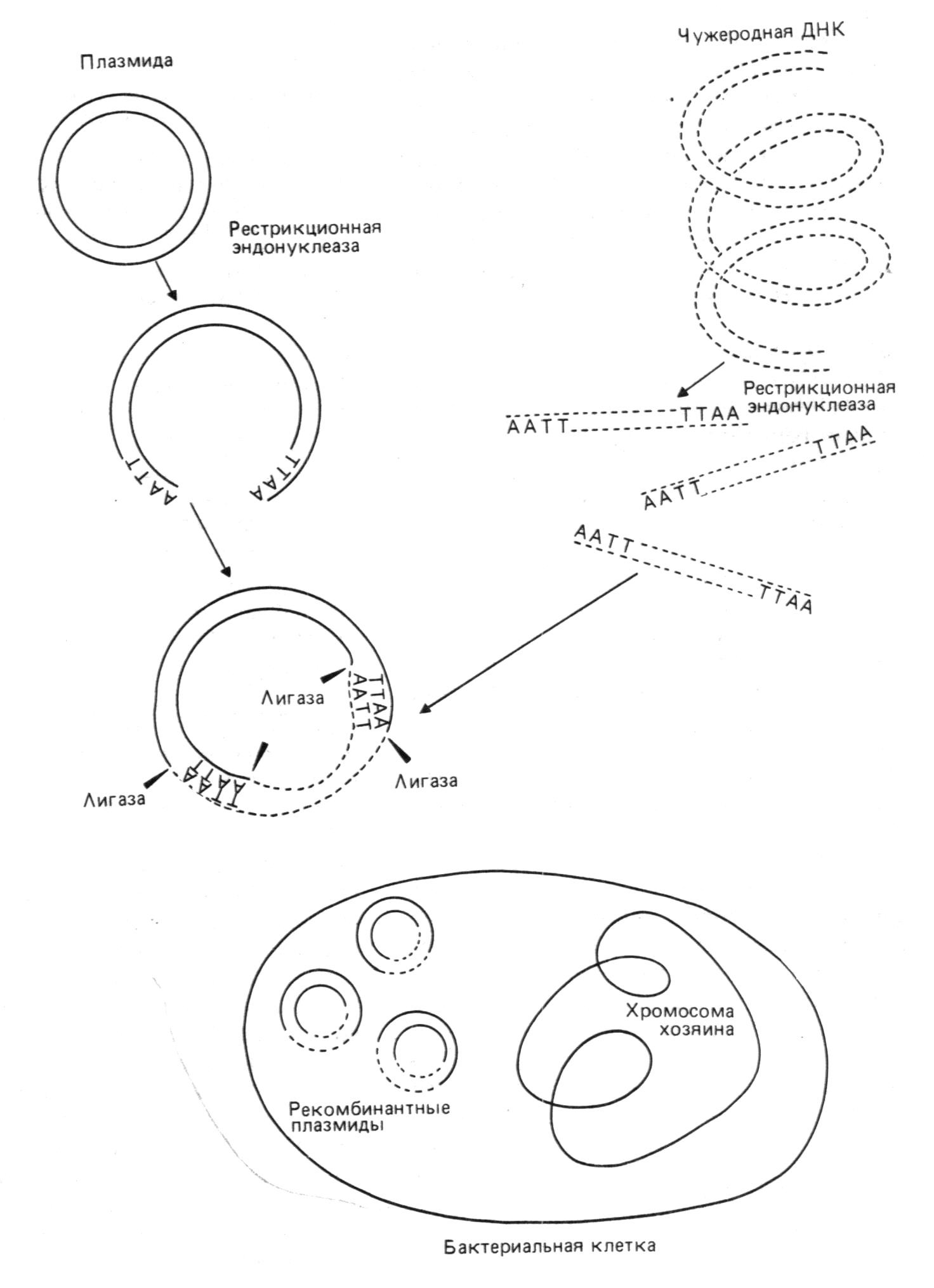
Программа общего назначения "COMMON" обеспечивает ввод последовательности в ЭВМ, а также проверку введенных данных в диалоговом режиме. Они позволяют также редактировать нуклеотидные последовательности: вводить замены и вставки, исключать нуклеотиды, вырезать, встраивать и объединять нуклеотидные последовательности и таким образом моделировать гибридные и мутантные молекулы ДНК. Ввод последовательностей в память машины можно осуществлять вручную с клавиатуры, но в последнее время созданы приборы для автоматического сканирования авторадиограмм и секвениру-ющих гелей, полученных при использовании флуоресцентных меток , передачи данных сразу в компьютер и последующего анализа последовательности с помощью специальных программ. В недавно вышедшей в издательстве IRL (Оксфорд) книге "Анализ сиквенса нуклеотидных кислот и белков" подробно описываются как конструкция сканирующих устройств, так и программы для чтения авторадиограмм. Созданы программы для восстановления первичных структур высокомолекулярных ДНК на базе данных сиквенса фрагментов, полученных при ее Неспецифическом расщеплении (например, ультразвуком,). Родство между любой парой фрагментов ДНК выявляется на основании совпадения последовательности нуклеотидов в их структурах, причем эти совпадающие последовательности и являются местом перекрывания и такие два фрагмента могут быть объединены в более протяженную структуру. Процесс отбора фрагментов и стыковки продолжается до. тех пор, пока не будет восстановлена вся первичная структура исследуемой ДНК. Одной из такого рода программ является "CONTIG" (существует ее вариант для компьютера IBM PC), созданная в лаборатории Ф.Сангера (Кембридж, Англия). Ниже приводятся основные операции, которые позволяет осуществлять программа "CONTIG":
1) хранение сиквенса каждого фрагмента;
2) отбор смежных фрагментов и сборка последовательностей из них;
3) сравнение данных, полученных при чтении новых авторадиограмм, с уже установленными последовательностями;
4) объединение двух фрагментов с помощью третьего, представляющего собой область перекрывания первых;
5) поиск участков ДНК, комплементарных уже установленным, что является проверкой правильности сборки полной структуры ДНК.
ЗАКЛЮЧЕНИЕ.
Метод Максама-Гилберта и метод Сэнгера основаны на одном принципе. В первом используется специфическое расщепление ДНК, обусловленное природой оснований, во втором - статистический синтез ДНК, заканчивающийся на каком-либо одном из 4 нуклеотидов. Таким образом, основой обоих методов является получение полного (статистического) набора фрагментов ДНК, оканчивающихся на каждом из четырёх нуклеотидов.
Химический метод (метод Максама-Гилберта) проще использовать в том случае, когда исследуемая ДНК не слишком велика (200-500 звеньев). В том случае, если речь идет о секвенировании высокомолекулярной ДНК, лучше применять метод полимеразного копирования (метод Сэнгера) , чтобы не вводить процедуру рестриктазного расщепления с выделением индивидуальных фрагментов. При энзиматическом секвенировании протяженных одноцепочечных ДНК (например, бактериофагов) можно применять набор олигонуклеотидов-затравок, синтез которых в настоящее время не требует больших затрат времени и труда. Для двутяжевых высокополимерных ДНК наиболее удобен метод слепого энзиматического секвенирования с применением универсальной затравки (их выпускают многие фирмы) и обработки данных с помощью ЭВМ. Химический метод также может быть применен, но в этом случае необходимо вырезать из вектора исследуемые фрагменты ДНК, и это усложняет всю процедуру.
СПИСОК ЛИТЕРАТУРЫ.
1. Жарких А.А. Методы филогенетического анализа генов и белков //Молек.биология. (Итоги науки и техники. ВИНИТИ; М. 1985)
2. Компьютерный анализ генетических текстов / А.А.Александров, Н.Н.Александров, М.Ю.Бородовский И ДР. М.: Наука.
3. Методы молекулярной генетики и генной инженерии. Отв. ред. Р.И.Салганник.-Новосибирск: Наука. Сиб. отд-ние, 1990.
4. Молекулярная клиническая диагностика.Методы: Пер. С англ. / Под ред. С.Херрингтона, Дж.Макги. – М.: Мир, 1999.
5. Шабарова З.А., Богданов А.А. Химия нуклеиновых кислот и их компанентов. – М.: Химия, 1978.
6. Шабарова З.А., Богданов А.А., Золотухин А.С. Химические основы генной инженерии: Учебное пособие . – М.: Изд-во МГУ, 1994.
7. Экспериментальные методы исследования белков и нуклеиновых кислот / Подред. М.А.Прокофьева. – М.: Изд-во МГУ, 1985.
Похожие работы
... генно-инженерных исследований. Многие из этих вопросов были подняты самими учеными активно работающих в данной области. В настоящее время большинство исследователей считали, что опасения касающиеся, генной инженерии, не имеют достаточно оснований, но многие этические проблемы остаются нерешенными и продолжают возникать новые. В прошлом генетика и медицинская генетика развивалась как относительно ...
... олигонуклеотидов—одну полуавтоматическую, а вторую в комплексе с компьютером. В 1982 г. цена этих приборов на американском рынке составляла 36000—39500 долл.[2]. К открытиям связанным с достижениями генной инженерии нужно прибавить то, что огромный генетический «чертеж» многоклеточного существа просчитан полностью. Я думаю это можно назвать достижением века. После восьми лет работы многих ...
... гистон), и от мыши. Вскоре аналогичная работа была выполнена в нашей стране группой специалистов под руководством С. И. Алиханяна и А. А. Баева. Достижения генетики и химии нуклеиновых кислот позволили разработать методологию генной инженерии: —открытие явления рестрикции — модификации ДНК и выделение ферментов рестриктаз для получения специфических ферментов; —создание методов химического ...
... через дыхательные пути человека). Воздушно- капельным путем (за счет образования стойких аэрозолей) распространяются многие респираторные инфекции (грипп, коклюш, дифтерия, корь, туберкулез и др.). Микробиологическая чистота воздуха имеет большое значение в больничных условиях (особо- операционные и другие хирургические отделения). Микрофлора человека и ее значение. Ребенок развивается в ...
0 комментариев