Навигация
Врожденные дефекты печени
56417
знаков
0
таблиц
0
изображений
5. Врожденные дефекты печени
К врожденным дефектам относятся аномалии развития печени (каверноматоз воротной вены, врожденные аномалии сосудов печени, артериопеченочная дисплазия) и желчевыводящих путей, а также различные нарушения метаболизма. Продукты обмена могут накапливаться в цитоплазме или лизосомах -болезни “накопления “ (гликогенозы, липидозы, гемохроматоз, холестеринозы и др.). Метаболические нарушения могут быть генетически обусловлены и связаны с энзимопатиями или структурными изменениями мембран (например, отсутствие рецептора). Врожденные дефекты клинически проявляются нарушениями обмена жиров, углеводов, липидов, желчных кислот, желчных пигментов.Так, гликогенозы характеризуют накоплением в гепатоцитах нормального гликогена или гликогена аномального строения. При нарушении метаболизма липидов (липидозах) в гепатоцитах и макрофагах накапливаются липиды и появляются своеобразные “клетки накопления” (болезнь Нимана – Пика, болезнь Гоше). Для генетически обусловленной недостаточности L1 – антитрипсина храктерно отложение аномального L1 – антитрипсина в перипортальных гепатоцитах. Гипофибриногенемии свойственно скопление фибриногена в эндоплазматической сети гепатоцитов. При гемохроматозе происходит накопление железа в сидеросомах гепатоцитов и макрофагов (гранулы гемосидерина), а также в просвете желчных капилляров. Скопление в гепатоцитах кристаллических включений и гранул, содержащих медь, наблюдается при болезни Вильсона – Коновалова.
МЕХАНИЗМЫ ПЕРВИЧНОГО ПОВРЕЖДЕНИЯ ГЕПАТОЦИТОВ
Альтерация гепатоцитов проявляется в виде дегенерации и некроза, развивающихся в условиях гипоксии, активации процессов ПСОЛ, мобилизации внутриклеточных протеолитических ферментов, в результате иммунных реакций.
Гепатотоксический эффект различных веществ определяется их сродством к рецепторному аппарату гепатоцита. Биологический фильтр печени – печеночные макрофаги ( клетки Купфера) элиминируют из крови эндотоксины или вирусы и, таким образом, предохраняют гепатоциты от повреждения.
Дегенеративные изменения гепатоцитов следует рассматривать как их повреждение, проявляющееся функциональными и морфологическими расстройствами. Морфологически наблюдается набухание клеток, уменьшение числа внутриклеточных органелл, дилятация цистерн эндоплазматического ретикулума, нарастающая дегрануляция цитоплазмы, уплотнение структуры органелл.
Необратимое нарушение целостности клеток (некроз) в зависимости от свойств гепатотоксических веществ, может быть очагового или диффузного характера. При продолжительных или повторных повреждениях печеночной паренхимы регенераторные способности гепатоцитов резко снижаются. При ограниченном повреждении определенной зоны выявляется перивенозный или перипортальный очаг некроза. Диффузные некрозы клинически проявляются нарушением функции печени, в первую очередь расстройствами обмена веществ (снижение способности к синтезу факторов свертывания крови, альбуминов, холинэстеразы и др.), падением активности реакций биологического трансформирования (переаминирование, трансаминирование и пр.). Выведение веществ через желчный шунт блокировано. В крови имеется гиперферментемия, гипербилирубинемия, холемия, накапливают ксенобиотики.
ГИПОКСИЯ печеночной паренхимы наступает в результате перфузионных нарушений и действия разобщающих ядов. Снижается интенсивность митохондриального окисления, истощаются энергетические запасы гепатоцитов. Угнетение окислительного фосфорилирования проявляется в замедлении транспорта ионов в мембране митохондрий, особенно кальция. В дальнейшем нарушаются функции других клеточных структур.
ПСОЛ активируется в результате образования активных форм кислорода в процессе биотрасформации субстратов. Гепатотоксичные ксенобиотики (например, хлороформ) способны образовывать свободные радикалы (СР.). До тех пор, пока сохраняется равновесие между образованием и элиминацией СР., повреждение клеток не происходит. Любые нарушения этого равновесия и увеличение концентрации СР. проявляются повреждением гепатоцитов. СР. обладают высокой реакционной способностью, они инициируют ПСОЛ мембран и непосредственно взаимодействуют с макромолекулами. Промежуточные продукты распада (альдегиды, перекиси, гидроксиальдегиды, кислоты, продукты распада трикарбоновых кислот) являются высокотоксичными веществами, так как, обладая СР., могут вызывать усиление процесса ПСОЛ или вступать во взаимодействие с макромолекулами белков. Токсичные СР. проявляют только локальное повреждающее действие, в то время как альдегиды, обладающие высокой диффузионной активностью, способны повреждать многие внутриклеточные структуры, в том числе и эндоплазматический ретикулум.
Накопление продуктов ПСОЛ в мембранах эндоплазматического ретикулума сопровождается общим снижением активности систем оксигеназ со смешанной функцией и деградацией терминального компонента этой системы – цитохрома Р450. В условиях патологии имеется обратимая корреляция в микросомальной фракции между содержанием цитохрома Р450 и концентрацией продуктов ПСОЛ.
Низкомолекулярные химические вещества (хиноны, эпоксиды) могут связываться с белками, образуя коньюгированные антигены. Это инициирует иммунный ответ гуморального или клеточного характера.
Нейтрализация токсических продуктов, образующихся в процессе ПСОЛ и имеющих в своем составе витамин Е, обеспечивается системой эндогенных антиоксидантов. Депонирование и перераспределение эндогенных антиоксидантов, в том числе и витамина Е, происходит преимущественно в печени. При ухудшении функционального состояния гепатоцитов показатели антиоксидантной активности липидов снижаются.
ФЕРМЕНТНЫЕ СИСТЕМЫ
Важнейшими изменениями в гепатоцитах при химическом поражении и гипоксии являются нарушения активности ферментативных систем мембран эндоплазматического ретикулума с резким снижением детоксикационной функции печени, снижение окислительного фосфорилирования в митохондриях, повышенная лизосомальная активность, накопление нейтральных липидов, угнетение белкового синтеза, дисбаланс ионного состава вследствие повреждения транспортных систем мембран, изменение активности внутриклеточных мессенджеров.
В основе изменений ферментативной активности гепатоцитов лежит резкое возрастание концентрации Са2+ цитоплазмы. При повышении уровня внутриклеточного Са2+ в первую очередь происходит активация фосфолипазы мембраны митохондрий и эндоплазматического ретикулума, ферментов гликогенолиза, растворимых протеинкиназ, кальмодулина.
При длительном воздействии этанола угнетается активность ферментов цикла Кребса, происходят выраженные изменения гепатоцитов, в первую очередь митохондрий, повышается мембранная проницаемость клеточных и внутриклеточных структур вследствие разрушения липидного матрикса из – за снижения в мембранах холестерина, являющегося стабилизатором липидного бислоя. При хроническом алкоголизме определяются гипертрофированные и атипические крипты, кристаллоидные отложения, повреждение и уменьшение текучести мембран вследствие уменьшения общего количества митохондриальных ФЛ. В гепатоцитах уменьшается содержание арахидоновой кислоты – основного субстрата ПСОЛ в мембранах.
Алкогольная интоксикация сопровождается повышением в плазме крови уровня свободных жирных кислот, образующихся на периферии. Усилению их синтеза способствует повышение коэффициента НАДН/НАД, которое происходит вследствие окисления этанола. Это, в свою очередь, ингибирует бета – окисление и усиливает эстерификацию холестерина.
Полагают, что под влиянием этанола и ацетальдегида в эндоплазматической сети, вероятно, синтезируются алкогольный гиалин, который состоит из белка, ФЛ, полисахаридов и обладает антигенными свойствами. Алкогольный гиалин и антитела циркулируют в крови, образуют иммунные комплексы, вызывая иммунокомплексное повреждение (подробно о влиянии алкоголя на функции печени см. соответствующий раздел)
Ртуть и ее органические соединения повреждают мембраны внутриклеточных структур: ядро, митохондрии, лизосомы, эндоплазматический ретикулум, блокируя сульфгидрильные группы белковых молекул, входящих в состав мембран.
ИНДИКАТОРЫ ПОВРЕЖДЕНИЯ ПЕЧЕНИ - ФЕРМЕНТЫ
Все метаболические процессы в печени осуществляются только благодаря содержащимся в гепатоцитах ферментам. Синтез ферментов – одна из важнейших функций печени, а динамическое постоянство ферментных констелляций в печени – необходимое условие ее нормального функционирования. Ферменты имеют белковую природу и синтезируются рибосомами. Вместе с тем, все клеточные органеллы обладают своим специфическим набором ферментов, определяющим их биологическую роль. Митохондрии содержат в основном ферменты энергетического обмена (ферменты окислительного фосфорилирования цикла Кребса, АТФазу и др.). С гранулярной эндоплазматической сетью связаны ферменты белкового синтеза, с гладкой ее частью – ферменты углеводного, липидного обмена, большинство реакций детоксикации, с лизосомами – основные гидролазы.
В процессе распада большинство ферментов подвергается протеолизу. Другой путь их разрушения – прижизненная термическая инактивация. Некоторые ферменты выделяются с желчью (щелочная фосфотаза, лейцинаминопептидаза, j – глутамилтранспептидаза) или с мочой ( амилаза).
В клинической практике ферменты разделяют по функции клеток печени и
их мембран, определяющих активность этих ферментов в сыворотке крови.
Выделяют следующие группы ферментов.
Секреторные ферменты синтезируются гепатоцитами и в физиологических условиях выделяются в кровь ( холинэстераза, церулоплазмин).
Индикаторные ферменты выполняют внутриклеточные функции (ЛДГ, АлАТ, АсАт, альдолаза и др.) В физиологических условиях их содержание в крови небольшое.
Экскреторные ферменты образуются в печени, в физиологических условиях выделяются с желчью (лейцинаминопептидаза, бета – глюкуронидаза, 5 – нуклеотидаза, щелочная фосфатаза).
По локализации ферменты подразделяют следующим образом:
а) универсально распространенные ферменты, активность которых обнаруживается не только в печени, но и в других органах – аминотрансферазы, фруктозо –1 – 6 – дифосфатальдолаза;
б) печеночноспецифические ( органоспецифические) – ферменты, активность которых выявляется только в печени, ( урокиназа, аргиназа, фруктозо – А-фосфатальдолаза, холинэстераза, сорбитдегидрогеназа, орнитинкарбамилтрансфераза и др.);
в) клеточноспецифические ферменты печени относят преимущественно к гепатоцитам, звездчатым ретикулоэндотелиоцитам или желчным канальцам (5 - нуклеотидаза, щелочная фосфатаза, j – глутамилтранспептидаза);
г) органоспецифические ферменты – маркеры определенных органелл гепатоцита:
· Цитоплазматические (АсАТ, АлАТ, ЛДГ, аргиназа, альдолаза, лейцинаминопептидаза, сорбитолдегидрогеназа, орнитинкарбамилтрансфераза);
· Митохондриальные (глутаматдегидрогеназа, сукцинатдегидрогеназа, малатдегидрогеназа, цитохромоксидаза, урокиназа);
· Лизосомальные ( кислые гидролазы – кислая фосфатаза, арилсульфатаза,
a –глюкозидаза, дезоксирибонуклеаза, b – глюкуронидаза, рибонуклеаза,);
· Микросомальные (глюкозо – 6 –фосфатаза );
· Рибосомальные ( холинэстераза, церулоплазмин);
· Эндоплазматические ( ферменты детоксикации и конъюгации).
Дегенерация и некроз гепатоцитов сопровождаются изменениями клеточных мембран, и в кровь высвобождаются индикаторные ферменты, которые являются маркерами повреждения.
Выделяют 4 основных патологических синдрома поражения печени
1. Синдром цитолиза обусловлен нарушением проницаемости мембран гепатоцитов и их органелл, приводящим к выделению составных частей клеток в межклеточное пространство и кровь. Для синдрома цитолиза характерны: повышение активности в крови ферментов – индикаторов цитолиза и печеночно – клеточных некрозов – АлАТ, АсАТ, альдолазы, глутаматдегидрогеназы, ЛДГ, и ее изоферментов ЛДГ – 4 и ЛДГ- 5; гипербилирубинемия; повышение в сыворотке крови концентрации витамина В12 и железа.
2. Синдром холестаза обусловлен нарушением желчевыделительной функции печеночных клеток с нарушением образования желчной мицеллы и поражением мельчайших желчных протоков. Синдром холестаза сопровождается повышением активности щелочной фосфатазы, лейцинаминопептидазы, гаммаглутамилтранспептидазы, 5 – нуклеотидазы; гиперхолестеринемией, повышением уровня фосфолипидов, бета - липопротеидов, желчных кислот, гипербилирубинемией.
3. Синдром печеночно-клеточной недостаточности отражает изменения основных проб печени, оценивающих поглотительно – экскреторную, метаболизирующую и синтетическую функции печени. Он включает:
а)печеночную (продукционную) гиперазотемию - повышение уровня сывороточного аммиака, фенолов, индикана, ароматических аминокислот, ( фенилаланина, тирозина, триптофана);
б) недостаточность синтетической функции печени – снижение уровня альбуминов, прокоагулянтов (II, V, VII факторов свертывания крови), протромбина, холестерина, падение активности холинэстеразы в сыворотке крови.
4. Иммуновоспалительный синдром обусловлен сенсибилизацией клеток иммунокомпетентной ткани и активацией ретикулогистиоцитарной системы. Для этого синдрома характерно: повышением уровня гамма – и бетаглобулинов, а также общего белка сыворотки крови; иммуноглобулинов А, G, М; появление неспецифических антител, в том числе к ДНК, к гладкомышечным волокнам, митохондриям; изменение количества и соотношения субпопуляций лимфоцитов (хелперов, супрессоров); изменение белково-осадочных проб (тимоловой, сулемовой, Вельтмана).
МАРКЕРЫ ИНФЕКЦИОННОГО ПОРАЖЕНИЯ ПЕЧЕНИ
Микроорганизмы могут быть обнаружены в гепатоцитах или клетках Купфера (включения цитомегаловируса, аденовируса, вируса желтой лихорадки, бактерий, токсоплазмы и т.д.) Независимо от возбудителя в печени возникают однотипные морфологические изменения, лежащие в основе реактивного гепатита и имеющие следующие характеристики:
а) различные виды дистрофий гепатоцитов (гидропическая, баллонная, жировая);
б) очаги некроза в различных отделах долек;
в) инфильтрация портальной и внутридольковой стромы различными клетками ( макрофагами, лимфоцитами, полиморфноядерными лейкоцитами);
г) образование гранулем, не имеющих специфического строения. Исключение представляют возбудители туберкулеза, проказы, сапа, которые вызывают развитие специфических гранулем.
ПЕЧЕНОЧНАЯ НЕДОСТАТОЧНОСТЬ
Все заболевания печени в зависимости от характера альтерации условно делят на следующие группы: гепатоцеллюлярные ( гепатиты, циррозы), холестатические, инфильтративные и опухолевые.
Вторичные патологические изменения являются общими для всех групп заболеваний. Поврежденные гепатоциты нарушают нормальный отток желчи. Непроходимость или воспаление желчных путей вызывают патологические изменения независимо от причины и характера поражения.
Печеночная недостаточность – это патологическое состояние всего организма, обусловленное недостатком нормально функционирующих гепатоцитов и характеризующееся комплексными нарушениями обмена веществ, дезинтоксикационной функции печени в сочетании с поражением мозга. При печеночной недостаточности нарушаются одна или несколько функций печени.
Острая (фульминативная) печеночная недостаточность – это следствие быстрой ( в течении нескольких недель или даже быстрее) потери печенью 90 или более процентов нормальных гепатоцитов вследствие их массивного некроза ( молниеносные формы острого вирусного или алкогольного гепатита, лекарственные поражения, пищевые и промышленные яды, сепсис, эндотоксикоз, вирусы герпеса, цитомегаловирус, Коксаки, вирус инфекционного мононуклеоза, острая гиповолемия, шок, гемолиз, острая надпочечниковая недостаточность, ожоги). При острой печеночной недостаточности в первую очередь падает утилизация печенью метаболитов, образуемых при осуществлении цикла лимонной кислоты, то есть лактата, пирувата, альфа – кетаглютарата. Эти метаболиты представляют собой органические кислоты, диссоциирующие во внеклеточной жидкости. В результате аккумуляции и диссоциации этих кислот во внутренней среде развивается метаболитический ацидоз, который путем падения ОПСС ведет к трудно устраняемой артериальной гипотензии.
Хроническая печеночная недостаточность развивается при поражениях печени аутоиммунного генеза, сердечной недостаточности, болезнях, сопровождающихся повреждением паренхимы печени (гепатиты, гепатозы, циррозы, опухоли, гельминтозы), генетических нарушениях обмена веществ (гликогенозы, галактоземия и др.); ЖКБ или опухолях, закрывающий общий желчный поток (в этом случае в результате накопления желчи в желчевыводящих путях повышается давление в желчных ходах и капиллярах, что ведет к нарушению секреции желчи гепатоцитами, ибо им теперь приходится преодолевать возросшее сопротивление; происходит массивное накопление компонентов желчи в печеночных клетках с их последующей гибелью и развитием соеденительной ткани – билиарный цирроз).
Выделяют два основных синдрома печеночной недостаточности:
1) Синдром холестаза
2) Синдром печеночно-клеточной недостаточности
Морфологической основой синдрома холестаза является застой желчи в желчевыводящих путях с последующей вторичной дистрофией гепатоцитов. Клинически характерны: кожный зуд, механическая (подпеченочная) желтуха, диспептические растройства, гепатомегалия.
Основой синдрома печеночно-клеточной недостаточности является первичная дистрофия гепатоцитов. Характерны сухость кожи, асцит, отеки, геморрагический диатез, портальная гипертензия, печеночно-клеточная желтуха, печеночный запах изо рта.
К осложнениям печеночной недостаточности относят печеночную энцефалопатию, крайней степенью выраженности которой является печеночная кома.
Общий патогенез печеночной недостаточности может быть представлен следующим образом. Повреждающий фактор вызывает изменение молекулярного строения мембраны гепатоцита с активацией ПСОЛ, что сопровождается деструкцией и повышением проницаемости мембран клетки и ее органелл. Выход лизосомальных гидролаз усугубляет повреждение мембран. Поврежденные печеночные макрофаги высвобождают фактор некроза и интерлейкин-1, в печени развиваются воспалительная и иммунная реакции. В организме синтезируются аутоантитела и появляются аутосенсибилизированные Т-киллеры, формируется аутоаллергическое повреждение гепатоцитов.
Формы печеночной недостаточности
1) Экскреторная (проявляется внутри и внепеченочным холестазом)
2) Васкулярная (портальная гипертензия и асцит)
3) Печеночно-клеточная (асцит, желтуха, энцефалопатия).
Метаболитические нарушения
при печеночно-клеточной недостаточности
Нарушение белкового обмена
Печень ответственна как за основание анаболические, так и за катаболические процессы обмена белков.
Синтез белков в печени осуществляется из свободных аминокислот. Это, прежде всего экзогенные аминокислоты, поступающие с кровью воротной вены из кишечника. Приток этих аминокислот в печень зависит от количественного и качественного состава пищи активности пищеварительных ферментов, фазы пищеварения и т.д. Эндогенные аминокислоты образуются в организме вследствие физиологического клеточного распада в других органах. Небольшое количество аминокислот образуется в самой печени из углеводов и жирных кислот.
Печень – место синтеза альбуминов, фибриногена, протромбина, проакцелерина, проконвертина, основной массы альфа и бета – глобулинов, гепарина. Синтез белков осуществляется в гепатоцитах рибосомами. Собственные белки и ферменты печеночных клеток синтезируются на свободных рибосомах и полисомах гиалоплазмы гепатоцитов, не связанных с мембранами эндоплазматической сети. Синтез белков «на экспорт» осуществляется рибосомами зернистой эндоплазматической сети.
Большинство заболеваний печени с тяжелыми повреждениями паренхимы сопровождаются снижением белково-синтетической функции гепатоцитов в результате угнетения каталитической активности мембраносвязанных ферментов и ферментативной активности субклеточных структур. Нарушается контакт рибосом с эндоплазматическим ретикулумом вследствие редукции мембран и уменьшения их белкового компонента.
Снижение белково-синтетической функции печени имеет следующие проявления:
1)Гипоальбуминемия, вследствие которой развивается гипоонкия, сопровождающаяся периферическими отеками, асцитом, гипотонией. Поскольку альбумины выполняют в организме антитоксическую (связывают метаболиты и ксенобиотики) и транспортную (связываясь с жирами, предотвращают возможность жировой эмболии, связываясь с билирубином, лишают его токсических свойств) функции, то токсичность эндо и экзотоксинов при гипоальбуминемии проявляется даже при их минимальной концентрации в плазме. Кроме того, известно, что альбумины участвуют в поддержании коллоидного состояния глобулинов крови, и последние легче выпадают в осадок (на этом основана проба Вельтмана, тимоловая проба).
2) Нарушение синтеза прокоагулянтов ведет к кровоточивости (этому так же может способствовать нарушение образования желчи, что вызывает затруднение всасывания жирорастворимого витамина К).
3) Снижение продукции транспортных белков ( трансферрина, переносящего ионы железа, церулоплазмина, переносящего ионы меди, цианокобаламина – ионы кобальта, транскортина, связывающего глюкокортикоиды и др.)
Расщепление белков до образования мочевины так же осуществляется в печени.
В гепатоцитах активно идут процессы утилизации аминокислот: их дезаминирование, переаминирование (трансаминирование) и декарбоксилирование. При значительных поражениях паренхимы, особенно при массивных никрозах, повышается уровень свободных аминокислот, остаточного азота в крови, при этом значительная часть аминокислот выделяется с мочой.
Нарушение реакций дезаминирования при патологи печени сказывается неблагополучно на состоянии организма, поскольку:
а) происходит усиленное выведение аминокислот с мочой, то есть организм бесполезно теряет необходимые для его жизнедеятельности вещества;
б) возрастает интенсивность декарбоксилирования аминокислот, что ведет к образованию биогенных аминов , например, гистамина;
в) усиливается интенсивность так называемых альтернативных путей их обмена, в ходе которых возможно образование токсических продуктов и даже обладающих канцерогенными свойствами ( некоторые продукты нарушенного обмена триптофана).
Для характеристики аминокислотного спектра крови определяют аминокислотное соотношение:
Вал + Лей + Изолей
_________________ = 3,0 – 3,5
Фен + Тир
При печеночной недостаточности это соотношение снижается.
Печень осуществляет катаболизм нуклеопротеидов с их расщеплением до аминокислот, пуриновых и пиримидиновых оснований. В печени последние превращаются в мочевую кислоту, выделяемую почками. Важно отметить, что конечные этапы катаболитических изменений белковых тел в печени одновременно представляют ее детоксицирующую функцию.
Нарушение углеводного обмена
Печень играет центральную роль в многочисленных реакциях промежуточного обмена углеводов. Среди них особенно важны описанные ниже процессы.
1) Превращение галактозы в глюкозу. Галактоза поступает в организм в составе молочного сахара. В печени происходит ее превращение в глюкозо-1- фосфат (Г-1-Ф). При нарушении функции печени способность организма использовать галактозу снижается (на этом основана функциональная проба печени с нагрузкой галактозой).
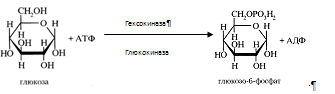
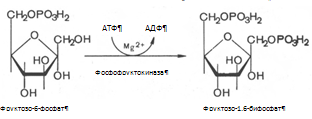
2) Превращение фруктозы в глюкозу Печень превращает фруктозу во фруктозо-1-фосфат (Ф-1-Ф) с помощью содержащейся в ней специфической фруктокиназы при участии АТФ. Ф-1-Ф расщепляется в печени альдолазой В.. Часть фруктозы под действием гексокиназы превращается во фруктозо-6-фосфат, промежуточный продукт основного пути распада глюкозы. Под действием глюкозофосфатизомеразы фруктозо-6-фосфат превращается в глюкозо-6-фосфат ( Г-6-Ф).
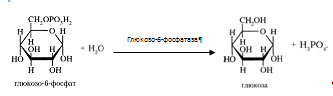
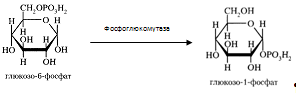
3) Синтез и распад гликогена Гликоген синтезируется из активированной глюкозы (Г-6-Ф). Печень может синтезировать гликоген и из других продуктов углеводного обмена, например, из молочной кислоты. Распад гликогена в печени происходит и гидролитически, и фосфоролитически. Под действием фосфорилазы образуется Г-1-Ф, который превращается в Г-6-Ф, последний включается в различные метаболитичекие процессы. Печень служит единственным поставщиком глюкозы в кровь, так как только под влиянием печеночной микросомальной Г-6-фосфатазы из Г-6-Ф освобождается глюкоза. Таким образом, под влиянием обратимых реакций распада и синтеза гликогена регулируется количество глюкозы в соответствии с потребностями организма. Уровень гликогена регулируется гормональными факторами: АКТГ, глюкокортикоиды и инулин повышают содержание гликогена в печени; адреналин, глюкагон, СТГ и тироксин - понижают.
4) Глюконеогенез. Глюкоза может синтезироваться из различных соединений неуглеводной природы, таких как лактат, глицерин, некоторые метаболиты цитратного цикла и глюкопластические аминокислоты (глицин, аланин, серин, треонин, валин, аспарагиновая и глютаминовая кислоты, аргинин, пролин, гистидин, оксипролин). Глюконеогенез связывает между собой обмен белков и углеводов и обеспечивает жизнедеятельность при недостатке углеводов в пище. При печеночной недостаточности в результате угнетения глюконеогенеза, снижения содержания гликогена в печени, угнетения реакции гепатоцитов на глюкагон, увеличения содержания в крови инсулина (вследствие уменьшения его инактивации печенью) возникает гипогликемия.
Таким образом, можно выделить следующие причины гипогликемии при печеночной недостаточности:
а) угнетение глюконеогенеза всей печенью из-за снижения числа функционально интактных гепатоцитов;
б) падение содержания гликогена в печени;
в) угнетение реакции гепатоцитов на эффект глюкагона как стимулятора глюконеогенеза;
г) рост содержания в крови инсулина как следствие падения его инактивации печенью.
5) Образование глюкуроновой кислоты. С обменом углеводов связан синтез глюкуроновой кислоты, необходимой для конъюгации плохо растворимых веществ (фенолы, билирубин и др.) и образования смешанных полисахаридов (гиалуроновая кислота, гепарин и др.)
В основе нарушений обмена углеводов при болезнях печени лежат повреждения митохондрий, которые ведут к снижению окислительного фосфорилирования. Вторично страдают функции печени, требующие расхода энергии, - синтез белка, эстерификация стероидных гормонов. Дефицит углеводов приводит также к усилению анаэробного гликолиза, вследствие чего в клетках накапливаются кислые метаболиты, вызывающие снижение рН. Следствием этого являются разрушение лизосомальных мембран и выход в цитоплазму кислых гидролаз, вызывающих некроз гепатоцитов. Нарушение углеводного обмена при патологии печени проявляются гипогликемией натощак вследствие истощения депо гликогена в печени, снижением способности организма поддерживать нормальный уровень глюкозы в крови.
Нарушение липидного обмена
Печень играет ведущую роль в обмене липидных веществ – нейтральных жиров, жирных кислот, фосфолипидов, холестерина. Участие печени в обмене липидов тесно связано с ее желчевыделительной функцией: желчь активно участвует в ассимиляции жиров в кишечнике. При нарушении образования или выделения желчи жиры в повышенном количестве выделяются с калом. Желчь усиливает действие панкреатической липазы и вместе с рядом других веществ участвует в образовании хиломикронов. Гепатоциты с помощью микроворсинок непосредственно захватывают липиды из крови. В печени осуществляются следующие процессы обмена липидов: окисление триглицеридов, образование ацетоновых тел, синтез триглицеридов (ТГ) и фосфолипидов, синтез липопротеидов, холестерина.
Гидролиз ТГ на глицерин и жирные кислоты происходит под действием внутрипеченочных липолитических ферментов. Печень является центральным местом метаболизма жирных кислот. В ней происходит синтез жирных кислот и их расщепление до ацетил-кофермента А, а так же образование кетоновых тел, насыщение ненасыщенных жирных кислот и их включение в ресинтез нейтральных жиров и ФЛ с последующим выведением в кровь и желчь. Катаболизм жирных кислот осуществляется путем бета - окисления, основной реакцией которого является активирование жирной кислоты с участием кофермента А и АТФ. Освобождающийся ацетил-кофермент А подвергается полному окислению в митохондриях, в результате чего клетки обеспечиваются энергией.
Кетоновые тела (ацетоуксусная, бета – оксимасляная кислоты и ацетон) образуются исключительно в печени. Возникающий в патологических условиях кетоз связан с диссоциацией кетогенеза в печени и утилизацией кетоновых тел в других органах. Из жирных кислот, глицерина, фосфорной кислоты, холина и других оснований печень синтезирует важнейшие составные части клеточных мембран – различные ФЛ. Синтез нейтральных жиров и фосфолипидов связан главным образом с митохондриями, а также с гладкой эндоплазматической сетью.
Синтез холестерина в основном происходит в печени и кишечнике. Он представляет собой важную составную часть плазмы крови и используется для синтеза кортикостероидных гормонов, витамина Д, желчных кислот и липидных структур мембран. Основная масса холестерина синтезируется гладкой эндоплазматической сетью. Уровень холестерина поддерживается постоянным в результате синтеза, катаболизма и выведения избыточного количества с желчью в кишечник; пятая часть его выделяется с калом, а большая часть всасывается вновь, обеспечивая печеночно-клеточную циркуляцию. Печеночные клетки полностью ответственны за удаление избыточного количества холестерина с желчью. Нарушение печеночно-клеточной циркуляции вследствие окклюзии желчевыводящих путей приводит к резкому возрастанию синтеза желчных кислот из холестерина.
Если гепатоцеллюлярные болезни снижают число нормальных гепатоцитов до определенного уровня, то падение синтеза холестерина в печени преобладает над снижением его экскреции в просвет кишечника таким образом, что в сыворотке крови падает концентрация холестерина.
Если внешние по отношению к печени системные растройства обмена веществ приводит к гиперхолистеринемии, то печень начинает выделять с желчью больше холестерина, и его концентрация в желчном пузыре растет. Рост содержания холестерина в крови предрасполагает к формирования камней желчного пузыря.
В печени происходит синтез липопротеидов, особой транспортной формы ФЛ.
При повреждении гепатоцитов синтез ФЛ в них угнетается и накапливаются нейтральные липиды, что ведет к жировой дистрофии печени, при которой содержание ТГ может достигать 80% массы печени. В основе жирового перерождения печени лежат процессы, которые приводят к недостаточности окслительно-восстановительных реакций, что сопровождается снижением содержания АТФ в гепатоцитах, либо ведут к прямому повреждению структуры печеночных клеток.
Среди причин можно выделить следующие:
1) Нарушение кровоснабжения печени по системе печеночной артерии (при патологии сердца, анемиях, снижении ОЦК и т.д.);
2) Гипоксемии различного генеза;
3) Инфекционные, вирусные поражения гепатоцитов;
4) Действие токсических веществ (четыреххлористый углерод, фосфорорганические вещества: хлорофос, карбофос, и др.; хлороформ и пр.);
5) Углеводное голодание (сахарный диабет, полное голодание или длительное малокалорийное питание), поскольку именно глюкоза является основным поставщиком молекул АТФ;
6) Снижение интенсивности утилизации в печени жира (например, при длительном действии алкоголя);
7) Нарушение синтеза в печени белков, в том числе составляющих белковую часть транспортных липопротеидных комплексов, в результате чего превалирует образование ЛПНП и ЛПОНП;
8) Избыточный синтез жиров из углеводов ( при чрезмерном употреблении углеводов, перекрывающем энергетические потребности организма);
9) Нарушение синтеза ФЛ. Известно, что ФЛ значительно более «водорастворимы», чем жиры. Они быстро покидают гепатоциты, поскольку активно используются для новообразования клеточных и субклеточных мембран. Для синтеза же ФЛ кроме глицерина и жирных кислот нужна фосфорная кислота и азотистые основания, для образования которых необходимы метильные группировки, донаторами которых являются метионин и холин. Вот почему на ранних этапах жирового перерождения печени показано назначение последних.
10) Все случаи длительной гипергликемии (алиментарной, транспортной, ретенционной), что сопровождается поступление избыточного количества жира в гепатоциты.
При поражении гепатоцитов ингибируется процесс эстерификации холестерина и синтез холестерина, поэтому накапливается уксусная кислота, являющаяся субстратом для его образования. В большом количестве уксусная кислота проявляет цитотоксическое действие. Роль желчных кислот в обмене холестерина значительна, поэтому различные нарушения метаболизма желчных кислот сопровождаются серьезными нарушениями обмена холестерина.
В крови при патологии печени содержание эфиров холестерина снижено, а уровень свободного холестерина повышен.
Известно, что в печени происходит детоксикация жирных кислот с короткой цепью (ЖККЦ), образующихся в кишечнике под влиянием бактериальной флоры ( бутановая, валериановая, капроновая и др.).
Нарушение функции печени сопровождается увеличением содержание не только ЖККЦ, но и жирных кислот с длинной цепью. Для головного мозга наиболее токсичнее бутановая и изовалериановая кислоты. ЖККЦ транспортируются альбумином, поэтому в условиях гипоальбуминемии ЖККЦ накапливаются в тканях мозга и синапсах. При избыточном образовании ЖККЦ связывающие способности альбуминов могут быть исчерпаны.
ЖКККЦ ингибирует синтез мочевины и активность глутаминовой дегидрогеназы (два основных пути утилизации аммиака), нарастает гипераммониемия. Они обладают способностью потенциировать токсическое действие аммиака, и их синергический эффект оказывается значительно выше. ЖККЦ оказывают прямое воздействие на нейронные и синаптические мембраны, блокируя транспорт ионов на мембране нейрона и, соответственно, проведение импульсов.
Нарушение обмена гормонов и витаминов
Стероидные гормоны (глюкокортикоиды, андрогены, эстрогены, альдостерон) образуются вне печени, но ей принадлежит важнейшая роль в их инактивации и распаде. Печень осуществляет ферментативную инактивацию и конъюгацию стероидных гормонов с глюкуроновой и серной кислотами, активно влияет на гомеостатическую регуляцию уровня глюкокортикоидных гормонов. Она так же синтезирует специфический транспортный белок крови – транскортин, который связывает гидрокортизон, делая его временно неактивным.
Инактивация серотонина и гистамина совершается путем окислительного дезаминирования с участием МАО и гистаминазы. Повышение концентрации гистамина может быть одной из причин кожного зуда и язвообразования в желудочно-кишечном тракте.
Печень участвует в обмене почти всех витаминов, в ней происходит их депонирование и частичное разрушение. Всасывание поступающего с пищей жирорастворимого витамина А в кишечнике вместе с другими веществами липидной природы происходит благодаря эмульгирующему действию желчи. Большая часть витамина А накапливается печенью в мельчайших жировых капельках в цитоплазме печеночных клеток и звездчатых ретикулоэндотелиоцитов. В печени провитамин А каротин превращается в витамин А.
При печеночной недостаточности нарушается всасывание в кишечнике, накопление в печеночной ткани и поступление витамина А в кровь. Присутствие желчи в кишечнике – необходимое условие всасывание и других жирорастворимых витаминов - Д, Е, К. Витамин Е (токоферол) ингибирует процессы окисления, и его недостаток в организме ведет к повреждению паренхимы печени. Витамин К участвует в синтезе факторов протромбинового комплекса, осуществляемом гепатоцитами, и недостаточное его всасывание в кишечнике служит одной из причин гипопротромбинемии и геморрагического диатеза при патологии печени.
Обмен большинства витаминов комплекса В непосредственно связан с функцией печени. Многие из них входят в состав коферментов. Функции окислительных дыхательных ферментов связаны, в частности, с присутствием в ткани витамина В1, депонируемого в форме кокарбоксилазы и участвующего в декарбоксилировании L-кетокислот. Витамин В2(рибофлавин) активно участвует в окислительном дезаминировании аминокислот. Витамин В5 (пантотеновая кислота) входит в состав ацетилкофермента А и непосредственно связан с последними этапами цикла Кребса в образовании конечных продуктов метаболизма белков, жиров, углеводов, детоксикацией ароматических аминов. Витамин В6 (пиридоксин) является коферментом ферментов, участвующих в трансаминировании и декарбоксилировании аминокислот, в катализе основных жирных кислот, входит в состав фосфорилазы, гистаминазы.
Нарушение обмена железа
В норме дневной рацион человека содержит около 10-20 мг железа, из которых всасывается 1-1,5 мг. Количество всосавшегося железа зависит от его запасов в организме: чем выше потребность, тем больше железа всасывается. Всасывание происходит в верхнем отделе тонкой кишки. В клетках слизистой оболочки железо находится в цитозоле. Некоторая его часть связывается и хранится в виде фермента, который впоследствии либо используется, либо теряется в результате слущивания клеток. Таким образом, ферритин-белок, депонирующий железо. Часть железа, предназначенная для метаболизма в других тканях, переносится через базолатеральную мембрану гепатоцита и связывается с трансферрином, основным транспортным белком железа в крови. Трасферрин является гликопротеином, синтезируемым в печени. Общая железосвязывающая способность сыворотки обусловлена трансферрином. В норме трансферрин насыщен железом примерно на треть. Физиологическое поглощение железа ретикулоцитами и гепатоцитами зависит от рецепторов трансферрина на клеточной поверхности, которые обладают сродством преимущественно к трансферрину, связанному с железом. Комплекс железа с рецептором входит внутрь клетки, где железо высвобождается. При насыщении клетки железом клеточные рецепторы трансферрина уничтожаются. Когда происходит полное насыщение трансферрина, железо циркулирует в формах, не связанных с трансферрином, в виде соединений с низкомолекулярными хелаторами. В такой форме оно легко поступает в клетки независимо от степени насыщения их железом.
Содержание железа в организме взрослого человека составляет 4-5г, из них 3 г – в составе гемоглобина, миоглобина, каталазы и других дыхательных пигментов или ферментов. Остальное железо депонируется.
Печень – основное место хранения железа, всасывающегося в кишечнике. При ее предельном насыщении железо откладывается в других паренхиматозных органах, включая ацинарные клетки поджелудочной железы и клетки передней доли гипофиза. Ретикулоэндотелиальная система становится местом преимущественного отложения железа лишь при его внутривенном введении. Железо из разрушенных эритроцитов накапливается в селезенке.
При небольших запасах железа, как уже было отмечено, оно хранится в виде ферритина. При избыточном поступлении в клетку железо откладывается в виде пигмента гемосидерина, который локализуется в лизосомах. Все повреждения печени, вызванные повышенным содержанием железа, получили общее название гемосидерозы (преимущественное накопление железа в ретикулоэндотелиальной системе).
Преимущественное накопление железа в паренхиматозных клетках – это гемохроматоз. Клиническое понятие сидерозов (болезней накопления железа) включает наследственный гемохроматоз и синдром гемохроматоза вследствие анемий, алкогольного цирроза, массивных трансфузий, хронического гемодиализа.
Возможны несколько механизмов повреждающего действия железа на печень. Под влиянием железа усиливается перекисное окисление мембран органелл, что приводит к нарушению функций лизосом, митохондрий и микросом, снижению активности цитохром - С - окидазы митохондрий. Нарушается стабильность мембран лизосом с выделением гидролитических ферментов в цитозоль. Перегрузка железом приводит к активации звездчатых клеток печени и усилению синтеза коллагена типа I, формируется фиброз и цирроз печени.
Нарушение обмена меди
С пищей за сутки в организм поступает 2-5 мг меди. Она всасывается в кишечнике, поступает в печень, где связывается с синтезируемым в печени церулоплазмином, циркулирует в крови, обязательно захватывается органами, которые в ней нуждаются, и экскретируется с желчью.
При наследственном дефекте синтеза церулоплазмина (болезнь Вильсона – Коновалова, или гепатоцеребральная дистрофия или гепатолентикулярная дегенерация) увеличена абсорбция меди в кишечнике и экскреция ее с желчью. При этом увеличивается содержание в крови и тканях свободной меди. Снижение или отсутствие активности церулоплазмина нарушает поступление достаточных количеств меди к ферментам тканевого дыхания, кроветворным органам; свободная медь накапливающая в тканях, блокирует SH – группы многих ферментов. Следствием недостаточного использования меди является депонирование ее в печени, мозге, почках, роговице. Складывается парадоксальная ситуация: нарушение биологических процессов из-за недостаточного количества меди и накопление меди в тканях с симптоматикой интоксикации металлом.
Церулоплазмин синтезируется исключительно в цитоплазме гепатоцитов вокруг ядра. Депонированная в печени медь вторично ингибирует синтез церулоплазмина, снижая и без того недостаточное его содержание.
Механизмы токсичности меди. Медь является прооксидантом, и ее накопление ведет к повышенной продукции свободных гидроксильных радикалов, которые вызывают повреждение мозга и печени. Это подтверждается снижением содержания в печени антиоксидантов (восстановленного глютатиона и витамина Е); увеличением циркулирующих продуктов ПСОЛ. Митохондрии печени могут играть главную роль в генерации свободных радикалов, и в тоже время они являются потенциальными мишенями действия оксидантов. Нарушение дыхательной цепи митохондрий и снижение активности цитохром – С – окидазы увеличивает продукцию свободных радикалов благодаря утечке электронов из дыхательной цепи. Повреждающее действие меди связано также с инактивацией ферментов митохондрий головного мозга.
Медь легко соединяется с SH – группами глутатиона и многих ферментов, участвующих в окислительно-восстановительных реакциях. Это приводит к энергетическому голоданию, к которому наиболее чувствительна ЦНС. Сходное повреждение имеется в печени с включением в митохондрии нерастворимой формы меди.
В начале заболевания при болезни Вильсона - Коновалова медь накапливается экстрализосомально, в цитозоле печеночных клеток. Медь, связанная с SH – группами цитозольных протеинов, затрудняет секрецию гепатоцитами белков и ТГ, что ведет к стеатозу. В дальнейшем медь перераспределяется из цитозоля в лизосомы гепатоцитов. Часть ее поступает в кровь. Медь, сконцентрированная в лизосомах, вызывает переокисление липидов и повреждение лизосомальных мембран с выходом кислых гидролаз. Наблюдается некроз гепатоцитов, развивается хронический гепатит и гемолитическая анемия. Усиленное накопление меди в печени приводит к фиброзу и циррозу.
Нарушение антитоксической функции печени
Детоксикация разнообразных веществ в печени осуществляется путем их биотрасформации, фагоцитоза и элиминации через желчный шунт. Печень участвует в обезвреживании ряда токсических продуктов клеточного метаболизма или веществ, поступающих извне. Детоксикации подвергаются вещества, образуемые микробами в кишечнике и через портальную систему, попадающие в печень. Это токсические продукты обмена аминокислот - фенол, крезол, скатол, индол, аммиак. В гепатоцитах происходит биотрансформация веществ благодаря процессам окисления, восстановления, гидролиза, метилирования, конъюгации и др. Реакции детоксикации осуществляются с помощью ферментов, связанных с гладкой эндоплазматической сетью и митохондриями. Они обладают выраженной активностью и относительно невысокой специфичностью. Это позволяет участвовать им также в процессах метаболизма таких эндогенных субстратов как гомоны, жирные кислоты, холестерин, желчные кислоты, простагландины, а также различных ксенобиотиков.
Существуют два механизма детоксикации:
1) монооксигеназные системы эндоплазматического ретикулума и сопряженные с ним реакции конъюгации; этот механизм включается при попадании в печень преимущественно липотропных соединений;
2) внемикросомальные механизмы, локализованные в цитозоле, митохондриях, лизосомах; активность этих механизмов проявляется преимущественно в отношении водорастворимых соединений.
В печени купферовскими клетками осуществляется фагоцитоз макромолекулярных соединений, продуктов деградации фибрина, старых поврежденных клеток крови, интерлейкинов, фактора некроза опухоли, других цитокинов.
Выделение метаболитов, коньюгатов, ксенобиотиков из гепатоцитов происходит главным образом через систему желчных ходов или после обратного всасывания через почки. Поскольку поверхность гепатоцитов, обращенная к желчным капиллярам, высокопроницаема для микромолекул большинства органических веществ, в желчи многие вещества содержатся в концентрациях, близких к таковым в крови. Однако такие вещества, как новокаинамид, гиппуровая кислот, большинство глюкуронидов и др. выделяются в желчь из гепатоцитов путем активного транспорта против градиента концентрации.
Микросомальные механизмы детоксикации
Совокупность метаболических превращений эндогенных субстратов и ксенобиотиков в гепатоцитах может быть представлена в виде двух фаз.
Похожие работы
... формы (киста, свищ, желтуха)• Виды операций. Основные вмешательства на поджелудочной железе при первичном хроническом панкреатите - резекции и операции внутреннего дренирования • Больным с желчнокаменной болезнью выполняют вмешательства, направленные на восстановление оттока жёлчи и панкреатического сока (холецистэктомия, холедохостомия, папиллосфинктеропластика).ДИАБЕТ САХАРНЫЙ Сахарный диабет - ...
... крови Ø УЗИ органов брюшной полости, биопсия печени, и другие методы – по показаниям Цирроз печени Цирроз печени – хроническое полиэтиологическое прогрессирующее заболевание печени, характеризующееся значительным уменьшением количества функционирующих гепатоцитов, нарастающим фиброзом, перестройкой нормальной структуры паренхимы и развитием в последующем печёночной недостаточности ...

... 3) гепатодепрессии (печёночно-клеточной недостаточности, малой недостаточности печени, недостаточности синтетических процессов); 4) воспаления; 5) шунтирования печени; 6) регенерации и опухолевого роста. При подозрении на конкретную патологию учитываются основные биохимические синдромы, характерные для данного заболевания. За основу берётся стандартная программа функционального обследования, но ...
... №_____ от__________ Зав. кафедрой к. м. н. Бегимбетова Р. С. методические разработки для преподавателей Тема: Амбулаторное ведение больных с хроническим гепатитом. Составила ассистент Калиева Г. А. АЛМАТЫ 1997 Г. Значение темы. ...
0 комментариев