ХАРАКТЕРИСТИКА
НАРКОТИЧЕСКИХ
АНАЛГЕТИКОВ
Характеристика
нового наркотического
анальгетика
просидол
Сенсометрия
Биохимические
(уровень
глюкозы в крови
)
ОБЩАЯ
ХАРАКТЕРИСТИКА
ОБСЛЕДОВАННЫХ
БОЛЬНЫХ
Применение
просидола в
качестве компонента
общей анестезии
Применение
просидола для
лечения послеоперационного
болевого синдрома
Применение
просидола для
лечения хронических
болевых синдромов
ПРИМЕНЕНИЕ
ТАБЛЕТИРОВАННОЙ
ФОРМЫ ПРОСИДОЛА
ДЛЯ ПРЕМЕДИКАЦИИ,
КУПИРОВАНИЯ
ПОСЛЕОПЕРАЦИОННОГО
И ХРОНИЧЕСКОГО
БОЛЕВЫХ СИНДРОМОВ
В качестве
компонента
наркоза просидол
применяют в
дозе 200-300 мкг/кг/ч,
для кардиохирургических
операций на
открытом
сердце-500-600 мкг/кг/ч
Навигация
ХАРАКТЕРИСТИКА НАРКОТИЧЕСКИХ АНАЛГЕТИКОВ
Проведение комбинированной общей анестезии с применением наркотического анальгетика просидола
200615
знаков
19
таблиц
2
изображения
1 ХАРАКТЕРИСТИКА НАРКОТИЧЕСКИХ АНАЛГЕТИКОВ
(ОБЗОР ЛИТЕРАТУРЫ)
1.1 Роль наркотических аналгетиков в анестезиологии (механизм действия, влияние на организм).
Арсенал средств для общей анестезии за последнее время значительно расширился, поскольку процесс создания новых препаратов стимулируется успехами исследователей, углубляющих наше понимание механизмов формирования боли, физиологии процессов, сопровождающих развитие общей анестезии (1, 2, 3). В настоящее время созданы лекарственные препараты, с заранее заданными свойствами, обеспечивающими тот или иной компонент защиты пациента от хирургического стресса (3, 13, 14). Естественные успехи в фармакохимии, фармакологии и токсикологии способствуют внедрению в клиническую практику все новых и новых как ингаляционных, так и неингаляционных анестетиков которые со временем либо заменяются новыми, более современными средствами, либо широко начинают использоваться в клинической практике (2, 3, 9). Тем не менее, до сих пор наблюдается устойчивая тенденция к преимущественному использованию неингаляционных способов общей анестезии (1, 3, 4, 9).
Общую анестезию, как известно, характеризуют несколько компонентов, основными из которых являются: амнезия, анальгезия, нейровегетативная блокада, миорелаксация, управление кровообращением и метаболизмом (4, 5).
С момента формирования понятий о компонентах общей анестезии эти представления прочно вошли в клиническую анестезиологию и способствовали разработке современных комбинированных методов наркоза (4, 5).
Аналгетический компонент общей анестезии в настоящее время обеспечивается в основном препаратами морфинового ряда: петидином, метадоном, рацеморфином, фентанилом, суфентанилом, альфентанилом и др (1, 3, 9). Анестезиологическая практика достаточно долго и в определенной степени успешно обходилась без этих препаратов, однако после эры безраздельного «господства» ингаляционных средств для наркоза, успехи фармакологии определили развитие и широкое внедрение в практику неингаляционных препаратов, с помощью которых в последующем были разработаны новые неингаляционные методы общей анестезии (1, 2, 3, 4, 5).
С определенностью можно сказать, что результаты исследованных механизмов анальгезии (при обеспечении этого компонента общей анестезии), стимулировали поиск и создание новых препаратов с определенными заданными свойствами, при чем исследователи, как правило, планировали создание средств, обладающих при максимальном анальгетическом эффекте действия минимумом побочных и токсических свойств, а также отсутствием привыкания (4, 6, 8, 13, 15).
Несмотря на успехи электроанальгезии, рефлексотерапии, других методов обезболивания, фармакотерапия боли не теряет своей актуальности. Проблема боли является одной из важнейших в медицинской науке, поскольку она имеет большое не только медико-биологичекое, но социально-экономическое значение (2, 3, 5, 15).
Открытие опиатных рецепторов и антагонистов наркотических аналгетиков, типа налоксона, в значительной степени увеличивало возможности изыскания новых анальгетиков и их антагонистов, изучение механизма анальгетического эффекта морфиноподобных веществ, а также выяснение причин психической и физической зависимости, возникающих под их влиянием (3, 17, 18,19).
В последние годы в создании новых анальгезирующих средств достигнуты определенные успехи. Весьма значительны они и в изучении механизмов действия анальгетиков (3, 20). Обнаружение простогландинов, энкефалинов, выяснение альгогенной и анальгетической роли этих и ряда других эндогенных веществ, а также открытие опиатных рецепторов создали новую основу для понимания механизмов боли и процессов обезболивания и стимулировали поиск новых анальгетических средств (3, 13).
По современным представлениям клеточные рецепторы, в том числе рецепторы нейронов, представляют собой белковые участки клеточной мембраны, селективно связывающие соответствующие биологически активные вещества (17, 18). При этом происходит изменение ее проницаемости, сопровождающейся передвижением ионов, а следовательно, перемещение электрических зарядов, изменяющих функцию и метаболизм клетки в целом (1, 2, 4, 17, 18, 20).
Разумеется возможны и иные принципы воздействия химических соединений на нейроны и другие клетки, примером чему может быть физико-химический способ действия общих и местных анестетиков (20, 21).
В последние годы рядом авторов (17, 18, 22) было доказано наличие в структурах ЦНС специальных рецепторов для некоторых нейротропных веществ, в том числе и для опиатных анальгетиков. Точкой приложения действия наркотических анальгетиков в ЦНС являются рецепторы опиатов (17, 20, 23, 24). Они расположены гетерогенно в разных структурах ЦНС и в некоторых внутренних органах, при этом наибольшая плотность опиатных рецепторов отмечается в афферентных путях проведения болевой чувствительности (например, в желатиновой субстанции спинного мозга, в некоторых ядрах таламуса) (17, 24, 25, 26, 27). Характерным для рецепторов опиатов является стереоспецифичность и обратимость связывания с наркотическими аналгетиками (17, 20). Антагонисты наркотических анальгетиков, имея больший аффинитет (число занятых веществом рецепторов) к рецепторам опиатов, вытесняют их и сами связываются с этими рецепторами (3, 4, 26). Однако, несмотря на появление в клинической практике новых синтетических анальгетиков, большое значение в практике продолжают иметь наркотические анальгетики (группа морфина и его аналогов) вследствие их непревзойденной способности к болеутолению (1, 14, 15, 28).
Классическим представителем наркотических анальгетиков - опиатов - является морфин, выделенный из опийного мака еще в 1803 году. Морфин - основной алкалоид опийного мака, где его содержание колеблется от 3 до 23% (3, 4, 24, 25).
В последние годы уточнены терминологические критерии, согласно которым болеутоляющие соединения, содержащиеся в соке опийного мака принято называть опиатами, вещества другого химического строения, близкие по фармакологическим эффектам к опиатам, - опиоидами или опиатоподбными веществами (25, 26, 29).
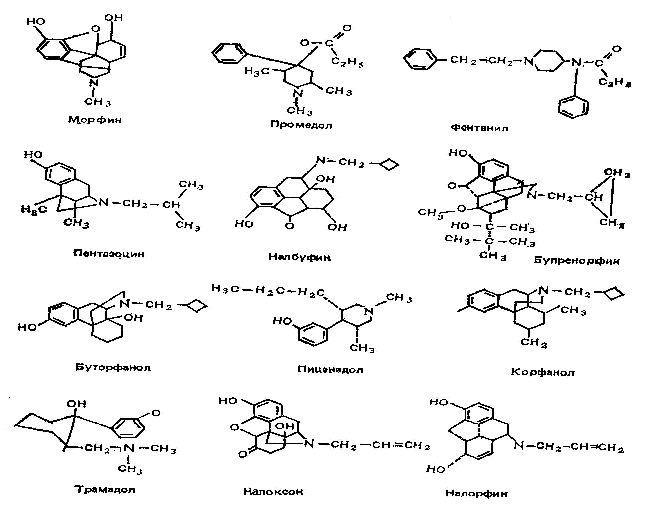
По избирательности и характеру их влияния на опиатные рецепторы наркотические анальгетики разделяются на несколько групп: морфиноподобные агонисты - морфин и его производные, промедол, фентанил и его производные, метадон, эторфин, трамадол; смешанные агонисты - антагонисты - пентазоцин, налбуфин, налорфин, корфанол; частичные (парциальные агонисты) - бупренорфин (1, 3, 29, 30, 31)
Формулы наркотических анальгетиков: (1, 3)
Нейрональные механизмы болеутоляющего действия наркотических аналгетиков и опиоидных пептидов
Современные представления о нейрофизиологии боли со всей очевидностью предопределяют, что аналгетический эффект опиатов и опиоидов представляет собой, по сути, результирующую их действия на различных уровнях центральной нервной системы (2, 4). Следует отметить, что появившиеся 6 - 8 лет назад данные о периферическом механизме их болеутоляющего действия не подтверждаются исследованиями последних лет, в которых убедительно показано, что морфин и фентанил не изменяют активность полимодальных ноцицептивных С-волокон ни при внутривенном введении, ни при аппликации на афферентные нервы (1, 32, 33, 34). В настоящее время следует считать общепризнанным, что передача ноцицептивных сигналов первично изменяется уже на уровне спинного мозга и что сегментарные механизмы действия опиатов и опиоидов играют существенную роль в реализации их болеутоляющего эффекта (34, 35, 36).
Избирательные влияния опиатов на ноцицептивные ответы соответствуют хорошо известным клиническим данным об уменьшении перцепции боли без изменения других сенсорных ощущений (34, 36, 37).
Угнетающее действие опиатов и опиоидных пептидов на нейрональную активность спинного мозга может реализоваться различными механизмами (29). Установлено, что значительное угнетение клеток V слоя под влиянием морфина происходит лишь тогда, когда он вводится в область желатинозной субстанции (1, 17, 34, 37, 38). При введении в желатинозную субстанцию- энкефалин, в отличие от морфина, угнетал ответы клеток V слоя не только на ноцицептивную, но и на неноцицептивную стимуляцию и даже уменьшал их спонтанную активность (1, 17, 36).
Болеутоляющий эффект опиатов и опиоидов при внутривенном введении может значительно варьировать, что, по-видимому, обусловлено различной ролью сегментарного компонента анальгезии у отдельных препаратов (24, 26). Так, бупренорфин, у которого при парентеральном введении анальгетическая активность в 1000 раз выше, чем у морфина, при интратекальном введении вызывает аналгезию, равную морфиновой (1, 4, 8, 38). Весьма вероятно, что болеутоляющее действие бупренорфина в большей степени по сравнению с морфином реализуется на супрасегментарном уровне, поскольку при микроинъекции в периакведуктальную область он вызывает анальгезию в 1000 раз большую, чем морфин (1, 8, 13, 36). Кроме того, значение сегментарного компонента в механизме болеутоляющего действия опиатов при субарахноидальном их введении должно оцениваться весьма осторожно, поскольку анальгезия в этих случаях возникает в результате достаточно высокой локальной концентрации опиатов и может варьировать у отдельных препаратов в зависимости от их липофильности, как было показано в отношении морфина и петидина (1, 36, 37, 39, 40, 41).
Болеутоляющее действие опиатов при системном введении обусловлено их влиянием как на сегментарном, так и супрасегментарном уровнях, причем в последнем случае изменяются влияния, регулирующие формирование восходящего ноцицептивного потока в спинном мозге (36, 39, 41, 42). О сегментарном действии системно введенных опиатов свидетельствуют данные о сохранении их анальгетического эффекта после перерезки дорсолатеральных канатиков спинного мозга или после полной спинализации. Показано, что морфин, промедол, фентанил, пентазоцин, дозозависимо и налоксонобратимо угнетали флексорный рефлекс и импульсную активность в вентролатеральных трактах спинного мозга (1, 41, 43).
Обычно аналгетический эффект опиатов и опиоидных пептидов объясняют их постсинаптическим действием (44, 45). Как показано с использованием метода внутриклеточной регистрации, опиаты и опиоиды вызывают гиперполяризацию мембраны как нейронов заднего рога спинного мозга, так и нейронов мезентериального сплетения и реснитчатого ганглия in vitro ( 1, 41, 45, 46). Кроме того, наркотические анальгетики и опиоиды налоксонобратимо устраняли деполяризацию и увеличение проводимости мембраны, вызванные глутаматом и ацетилхолином (45, 46, 47, 48). С другой стороны, уже сам факт избирательного угнетения ответов клеток заднего рога спинного мозга на ноцицептивные стимулы дает основание полагать, что оно обусловлено влиянием анальгетиков и на пресинаптическом уровне. Кроме того, известно, что опиатные рецепторы в спинном мозге локализуются не только пост - , но и пресинаптически ( 1, 48, 49, 50).
Однако данные об электрофизиологических проявлениях пресинаптического действия опиатов, лежащего в основе их болеутоляющего эффекта, довольно противоречивы, что в первую очередь определяется разнообразием представлений о пресинаптических механизмах формирования восходящего ноцицептивного потока на сегментарном уровне (50, 51, 52, 53) .
Болеутоляющее действие опиатов и опиоидных пептидов реализуется и на супрасегментарном уровне за счет нарушения проведения возбуждения по диффузной афферентной системе на уровне таких ее основных релейных звеньев, как ретикулярная формация ствола головного мозга и среднего мозга (52, 53, 54, 55).
ноцицеп-нисходя-структур
Принципиальным механизмом специфического болеутоляющего действия опиатов и опиоидов следует считать угнетение нейронов, связанных с ноцицептивной афферентацией, и снижение их активации через конвергирующие на них высокопорговые ноцицептивные входы (46, 49). Так, электрофоретическое подведение морфина и метэнкефалина всегда стереоспецифически и налоксонобратимо угнетает активность нейронов голубого пятна, которое имеет большую плотность опиатных рецепторов и участвует в регуляции болевой чувствительности (1, 3, 46, 54, 55, 56, 57).Можно считать, что в последние годы в целом сформулировано представление о болеутоляющем действии опиатов и опиоидов и достигнуто общее понимание излишней категоричности существовавших ранее контраверсий об исключительно сегментарных или супрасегментарных механизмах опиатной аналгезии (56, 57, 58, 59, 60, 61). Предложено достаточно много функциональных схем, согласно которым эта анальгезия формируется как за счет непосредственного угнетения релейных нейронов спинного мозга и особенно их активации через высокопороговые ноцицептивные входы, так и в результате усиления опиатами нисходящего торможения с различных антиноцицептивных структур центральной нервной системы (1, 55, 56, 57, 58, 59)
Влияние опиатов и опиоидов на аналгетические системы головного мозга
Одним из наиболее дискуссионных и наиболее уязвимых мест гипотезы о реализации опиатной анальгезии через эндогенные болеутоляющие системы является положение о способности опиатов усиливать нисходящее торможение и о способах этого усиления с учетом общеизвестных данных об отсутствии у них непосредственного активирующего действия на нейрональную активность (60, 62, 63, 64, 65).
Большинство гипотез о способности анальгетиков усиливать нисходящее торможение базируется на известной общности проявлений стимуляционной и опиатной анальгезии, хотя эта общность не позволяет судить об их тождественности по сути (1, 66, 67). Кроме того, в электрофизиологических исследованиях отправной точкой интерпретации действия опиатов на нисходящее торможение невольно служило допущение об идентичности нисходящего контроля сегментарных флексорных рефлексов и активности сегментарных нейронов, формирующих восходящий нецицептивный поток (3, 66, 67). Поэтому неудивительно, что некоторые данные о действии опиатов не укладываются в рамки существующих гипотез (1, 3, 55, 56, 68). Так, было показано, что морфин в минимальных анальгетических дозах в большей степени угнетал ответы нейронов V слоя и активность в антеролатеральных столбах спинного мозга, вызванные брадикинином (1, 55, 56, 69).
В работах последнего времени не находит подтверждения и устоявшееся представление о реализации нисходящего торможения, усиливаемого морфином ,через дорсолатеральные канатики спинного мозга (1, 55, 56, 70).
Дискуссионность гипотезы об усилении опиатами нисходящего торможения не уменьшилась и после того, как была предложена ее новая модификация (1, 70, 71), в которой специально сделан акцент на то, что опиаты и эндогенные опиоиды не активируют нейроны антиноцицептивных зон головного мозга, а уменьшают в этих зонах тормозные процессы, т. е. оказывают растормаживающее действие, которое проявляется как усиление нисходящего торможения (1, 3, 70, 72, 73).
Принципиально новое объяснение формированию опиатной анальгезии дали Д. Ле Барс и соавт. (1983, 1987) (1). Их модель регуляции болевой чувствительности предполагает, что ее усиление (гиперальгезия) или угнетение (гипоальгезия) обусловлены фармакологической модуляцией активности нейронов спинного мозга которые высокочувствительны к морфину в малых дозах, еще не влияющих на передачу ноцицептивных сигналов на сегментарном уровне (1). Согласно их гипотезе, боль возникает тогда, когда создается определенная контрастность между сигналами ноцицептивных и неноцицептивных нейрональных пулов, запускающих процессы перцепции боли. Опиатная аналгезия может возникать в результате уменьшения контрастности восходящего сигнала двумя способами: 1) за счет усиления исходной неноцицептивной активности нейронов, которое происходит под влиянием опиатов в малых дозах 2) за счет непосредственного угнетения опиатами в больших дозах передачи ноцицептивной информации на сегментарном уровне (1). Суммация супрасегментарного и сегментарного эффектов опиатов приводит к большему уменьшению контрастности сигналов и, следовательно, к усилению анальгезии (1).
Представленная схема формирования болеутоляющего действия морфина, несмотря на ее новизну и привлекательность, как и любая другая, не может объяснить все многообразие проявлений феномена опиатной анальгезии (73, 74, 75, 76). Неясно, как впишутся в рамки этой схемы болеутоляющие свойства других опиатов и опиоидных пептидов, отличающихся большей селективностью к определенным типам опиатных рецепторов (75, 76, 77).
Таким образом, вся совокупность полученных данных свидетельствует о том, что болеутоляющее действие разных опиоидов опосредуется различными механизмами и уровнями центральной нервной системы (1, 75, 76, 78, 79). Более того, можно считать, что каждый агонист определенного типа опиатных рецепторов имеет своеобразную, присущую только ему, последовательность вовлечения нейрональных субстратов и нейрофизиологических механизмов, формирующих его болеутоляющее действие (79, 80, 81, 82). Весьма вероятно, что отдельные процессы опиоидергической регуляции боли реализуются через различные типы опиатных рецепторов и что m-, б- и c - рецепторы в неодинаковой степени и весьма дифференцированно включаются в механизмы, модулирующие ноцицептивную информацию на супра- и сегментарном уровнях, определяющие интенсивность восходящего болевого потока и его нисходящую регуляцию (1, 79, 80, 81, 82, 83).
Существенное значение в формировании опиатной анальгезии имеет изменение восходящих влияний эндогенных анальгетических зон головного мозга (83, 84, 85,86).
Нейрохимические основы болеутоляющего действия аналгетиков и опиоидных пептидов.
Серотонинергические механизмы болеутоляющего действия опиатов и опиоидов.
Роль серотонинергических механизмов в реализации опиатной анальгезии активно исследуется уже в течение нескольких лет (1, 48, 50, 86,87). Казалось бы, что в этом вопросе достигнута определенная ясность, поскольку в виде общепринятой схемы считалось, что все воздействия, усиливающие серотонинергическую медиацию, увеличивают морфинную аналгезию, а угнетающие - ее ослабляют (1, 84, 85). В работах О. Berge и соавт. (1983), S. Dennis и P. Melzack (1980) обобщены основные данные об изменениях морфиновой анальгезии, определяемой в различных экспериментальных тестах, при изменении серотонинергической медиации (1).
Предполагается, что серотонинергическая медиация играет существенную роль в реализации анальгетического действия морфина только на супрасегментарном уровне и не имеет существенного значения в том компоненте его анальгетического эффекта, который обусловлен воздействием на спинной мозг (1, 56, 57,76) .
Ускорение синтеза серотонина под влиянием морфина может быть обусловлено и повышением уровня триптофана, поскольку морфин изменяет соотношение связанного и свободного триптофана в плазме и увеличивает содержание серотонина (1, 76, 77, 78). Весьма интересное предположение высказали F. Godefroy и соавт. (1980), согласно которому морфин может активировать синтез серотонина за счет воздействия на опиатные рецепторы (41, 56, 57, 82). Они полагают, что в этом процессе есть опиоидергическое звено, поскольку увеличение синтеза серотонина из триптофана блокировалось налоксоном и в то же время налоксон практически не изменял уровень триптофана в головном мозге (1, 41, 42, 78, 86). Несмотря на то, что неоднократно было показано ускорение кругооборота серотонина под влиянием морфина, очень мало обращалось внимания на два весьма существенных для интерпретации этих данных обстоятельства. Во-первых, во всех исследованиях морфин вводился, как правило, в одной дозе и, как правило, в сверханалгетической, хотя известно, что существует прямая зависимость между концентрацией морфина в головном мозге и его аналгетическим эффектом. Во-вторых, практически не исследовались зависимость и корреляция биохимических эффектов морфина от его уровня в центральной нервной системе (1, 3, 41, 56, 57, 58, 60).
Однако не все данные укладываются в рамки, казалось бы, хорошо обоснованной гипотезы об участии серотонинергических систем в морфиновой анальгезии (1, 41, 57, 58, 60, 80). Установлено, что разрушение большого ядра шва, селективная блокада нисходящих серотонинергических путей интратекальным введением 5,7-дигидрокситриптамина или блокада сегментарных серотониновых рецепторов снижает только исходную болевую чувствительность и в меньшей степени ослабляет болеутоляющее действие морфина (39, 40, 41, 43). В качестве доказательства участия серотонинсодержащих ядер шва в реализации морфиновой аналгезии обычно приводились данные об ее ослаблении при их электролитическом разрушении. Однако сейчас эти доказательства следует рассматривать весьма критически, поскольку при таком методе происходит деструкция нейрональных элементов различного нейрохимического профиля (1, 41, 43).
С гипотезой об усилении опиатами нисходящего торможения не согласуются также данные последнего времени о влиянии морфина на серотонинсодержащие нейроны ствола головного мозга (1, 4, 41, 57, 58). Если морфин действительно увеличивает это торможение, то он должен усиливать нейрональную активность нейронов - источников нисходящих проекций. Однако и ранее уже было известно, что морфин при системном введении или при микроинъекциях в ядра шва не оказывал однонаправленного действия на импульсную активность этих клеток (77, 78). Не исключено, что взаимодействие морфина с опиатными рецепторами надсегментарных структур сопровождается усилением выброса другого медиатора (вероятно, норадреналина), который воздействует в спинном мозге на серотонинергические терминалии и усиливает выход серотонина (1, 77, 78).
Роль серотонинергической медиации может быть разной в реализации болеутоляющего действия агонистов различных опиатных рецепторов. Анальгетическая активность метадона, меперидина и кодеина не изменялась при разрушении ядер шва среднего мозга, приводящем к снижению уровня серотонина в головном мозге (1, 41, 77, 78). Кроме того, было показано, что влияние морфина, леворфана, петидина, пентазоцина на содержание серотонина в синаптосомах головного мозга не коррелирует с их болеутоляющей активностью. Большинство из этих препаратов не препятствовали снижению содержания серотонина в мозге, вызываемому фенфлюрамином (50, 51, 77, 78). Следовательно, их болеутоляющее действие причинно не связано с изменением содержания серотонина в головном мозге (1, 3, 41, 51, 52). Эти данные дают основание полагать, что серотонинергическая медиация имеет ключевое значение в возникновении анальгетического действия c - агонистов и играет меньшую роль в болеутоляющем эффекте m - агонистов (1, 41). В связи с этим закономерно встает вопрос, насколько действительно специфичны для анальгетического действия опиатов изменения в серотониновой медиации (1, 41, 77, 78). Несомненно, что серотонин необходим для проявления центрального действия морфина, поскольку воздействия, изменяющие серотонинергическую медиацию, существенно влияют не только на его анальгетическое действие, но и на гипотермический, локомоторный, эйфоригенный эффекты (78, 79, 80). Однако вопрос о том, насколько серотонинергическая медиация специфически вовлекается в реализацию болеутоляющего эффекта опиатов, требует дальнейшего экспериментального обоснования (1, 41, 77, 78, 80).
Адренергические механизы болеутоляющего действия опиатов и опиоидов
В настоящее время имеются веские доказательства сопряженного участия опиоидергических и адренергических механизмов в регуляции болевой чувствительности. В исследованиях (88, 89) выявлено усиление болеутоляющего эффекта при совместном применении наркотических анальгетиков и адренопозитивных соединений, подтвержденное в других работах (1, 41, 88, 89). Вместе с тем уже на протяжении ряда лет дискутабельным является вопрос об уровнях и рецепторных основах опиоидадренергического взаимодействия (1, 41, 88, 89).
Вполне обоснованной представляется гипотеза о том, что угнетение ноцицептивных реакций под влиянием опиатов и опиоидов обусловлено усилением процессов нисходящего торможения структур спинного мозга, которое реализуется через норадренергические системы (83, 88).
Однако, несмотря на общепризнанную точку зрения о важном вкладе сегментарных структур в реализацию анальгетического влияния опиатов и опиоидов, весьма противоречивы данные о возможности взаимодействия собственно спинальных опиоидергических и адренергических систем (88, 89, 90). С одной стороны, о такой возможности прямо свидетельствуют результаты, показывающие развитие отчетливой анальгезии при одновременном введении морфина и адренергических агонистов в субанальгетических дозах под оболочки спинного мозга (88, 90, 91, 92). С другой стороны, с применением блокаторов разных нейромедиаторных систем спинного мозга установлено, что на этом уровне центральной нервной системы опиоидергические механизмы тесно связаны с серотонинергическими, но не адренергическими процессами (88, 89, 90, 93). He вносят ясности в этот вопрос данные об изменениях уровня и обмена норадреналина на фоне опиатной аналгезии (1, 41, 88, 90). Так, одни авторы обнаружили увеличение концентрации в спинном мозге одного из метаболитов норадреналина - З-метокси-4-оксифенигликоля - под влиянием морфина, а другие выявили противоположные или двуфазные сдвиги в его содержании (1, 41, 57, 58, 75, 76).
He менее противоречивы представления о рецепторных основах болеутоляющего эффекта, развивающегося при сочетании введения агонистов опиатных и адренергических рецепторов (87, 88, 89). Предполагается, что взаимодействие между опиоидами и адренопозитивными средствами не реализуется через общий рецептор, поскольку аналгетический эффект и тормозное действие на ноцицептивные ответы нейронов заднего рога спинного мозга не устраняются налоксоном (57, 58, 75, 76, 88, 90, 91). Авторы считают, что адрено- и опиатные рецепторы могут взаимодействовать и модулировать ноцицептивную передачу в спинном мозге (1, 41, 57, 58, 88, 89).
Наиболее убедительным и в значительной мере примиряющим противоречивые мнения является предположение о том, что взаимодействие адренергических и опиоидергических механизмов может осуществляться через разные рецепторы, но с последующим запуском общего аналгетического механизма (89, 92). Правомочность такого предположения подтверждается данными, показывающими, что опиаты угнетают высвобождение норадреналина из пресинаптических окончаний и что на уровне головного мозга адреномиметики и опиаты регулируют активность одних и тех же нейронов, но через разные рецепторы (1, 41, 55, 57, 56, 88, 90, 92, 93. 94). Аналогичным образом взаимодействие этих систем осуществляется и на сегментарном уровне, поскольку установлено, что анальгезия развивается при избирательной активации адренергической и опиоидергической систем спинного мозга (54, 55, 57, 77, 78, 88, 92).
С этих же позиций объясняют существенную роль адренергических механизмов в формировании зависимости и в реализации проявлений абстинентного синдрома (41, 78, 79, 88, 92). Считают, что существует общий механизм пресинаптической регуляции норадренергической передачи возбуждения в центральной нервной системе, в который вовлечены опиатные рецепторы, их активация угнетает высвобождение нейромедиаторов. Поэтому адренопозитивные средства и опиаты действуют через независимые друг от друга места связывания, хотя при этом может запускаться общий механизм, обусловливающий коррекцию повышенного оборота норадреналина при отмене опиатов (44, 45, 55, 56, 57, 88, 92, 93).
Необходимо подчеркнуть, что, несмотря на противоречивость представлений о механизмах опиоидадренергического взаимодействия, сам факт синергичного функционирования этих болеутоляющих систем не вызывает сомнения (1, 41, 88, 92, 95). В свою очередь, очевидно, его прикладное значение, заключающееся в возможности уменьшения дозировки наркотических анальгетиков и, следовательно, их побочных эффектов при сочетанном применении опиатных и адренопозитивных препаратов. Важным практическим аспектом является также пролонгация медикаментозного обезболивания адренопозитивными средствами у пациентов с толерантностью к опиатам и опиоидам (1, 41, 45, 55, 56, 57, 77, 78, 79, 88).
Морфин
Большой опыт клинического применения морфина показал, что препарат обладает несомненными достоинствами: обеспечивает глубокое обезболивание, не сопровождающееся амнезией, не вызывает сенсибилизации миокарда катехоламинами, не нарушает регуляции кровотока в головном мозге, сердце, почках, не оказывает токсического воздействия на печень, почки и др. (1, 3, 41, 103). Вместе с тем морфин нельзя признать идеальным анальгетиком прежде всего в связи с его высоким наркогенным потенциалом, способностью угнетать дыхание, вызывать обстипацию и некоторыми другими свойствами (1, 41, 96, 97, 98, 104, 106, 107, 108). Внутримышечное введение морфина обеспечивает оптимальную длительность действия морфина, тогда как после его внутривенного введения период полувведения (Т 1/2) составляет около 100 минут (1, 41, 97, 98). Морфин частично связывается с белками плазмы. Пороговое анальгетическое действие развивается при концентрации свободного морфина в плазме крови 30 нг/мл. Лишь незначительная часть от введенного морфина (менее 0,01%) обнаруживается в ткани головного мозга, что вероятно связано с относительно низкой липоидотропностью препарата. Выводится морфин из организма главным образом через почки преимущественно в виде глюкуронида. В экспериментах установлено, что активность морфина может изменяться в 7 раз в зависимости от времени суток и фазы менструального цикла у женщин (1, 41, 97, 98, 99, 100, 101, 106, 107).
Высокий наркогенный потенциал ограничивает длительное (за исключением инкурабельных больных) применение морфина (101). К сожалению, уже при его 1-2-кратном введении проявляется большое число побочных реакций, среди которых наиболее выражены угнетение дыхания, тошнота и рвота, спазмы гладкомышечных органов (101, 102). Примечательно, что негативные эффекты морфина прямо коррелируют с его концентрацией в крови (1, 104, 105, 106). На основании клинических наблюдений в ряде исследований было показано, что оптимальной однократной дозой морфина является доза 10 мг на 70 кг массы тела пациента, поэтому эта доза принята в качестве эталона для оценки других существующих и изучаемых болеутоляющих средств ІТаблица 1І (1, 4, 41).
Таблица 1
Сравнительная активность анальгетиков (по отношению к 10 мг морфина)
название препарата доза
| Кодеин | 90 мг | |
| Петидин | 75 мг | |
| Оксиморфин | 65 мг | |
| Пентазоцин | 50 мг | |
| Налбуфин | 30 мг | |
| Морфин | 10 мг | |
| Декстраморамид | 5 мг | |
| Бутарфанол | 2 мг | |
| Бупренорфин | 0,3 мг | |
| Фентанил | 0,1 мг |
Болеутоляющий эффект морфина обусловлен его влиянием на межнейронную передачу ноцицептивных (болевых) импульсов на различных уровнях центральной нервной системы (1, 102, 103)
Появились работы, где в противоположность общепринятым представлениям высказывается мысль о том, что опиоиды вызывают анальгетический эффект отчасти за счет взаимодействия с периферическими рецепторами (102, 103, 104).
Действие морфина, как известно, всегда сопровождается угнетением дыхания в той или иной степени, которая проявляется в уменьшении частоты, глубины дыхания, МОД и снижением чувствительности дыхательного центра к двуокиси углерода (99, 100, 109). В связи с гиповентиляцией отмечается накопление двуокиси углерода в альвеолярном воздухе и крови, приводящая к развитию дыхательного ацидоза (99, 100, 110). Гиперкапния и падение насыщения церебральной крови кислородом в свою очередь приводит к расширению сосудов головного мозга и повышению внутричерепного давления. Морфин также значительно изменяя функцию сердечно-сосудистой системы, вызывает гипотензию, снижает сердечный выброс, угнетает атриовентрикулярную проводимость в 33% случаях (99, 100, 111, 112). Кроме того, общеизвестно, что морфин вызывает тошноту в 20-40% и рвоту в 10-15% случаев, в 60-85% случаев он вызывает головокружение. Морфин и другие наркотические анальгетики угнетают также кашлевой рефлекс (101, 102, 104, 105, 111, 112). Сравнительная характеристика активности наркотических анальгетиков приведена вІ Таблице 2І (4).
Таблица 2
Сравнительная активность некоторых анальгетических препаратов по отношению к морфину, сила которого приравнена к 1
| Степень анальгезии | Препарат | Сила |
| Очень сильная | Суфентанил | 1000 |
| Фентанил | 100-300 | |
| Бупренорфин | 40-50 | |
| Альфентанил | 10-50 | |
| Оксиморфон | 12-15 | |
| Сильная | Бутарфанол | 8-11 |
| Гидроморфон | 7-10 | |
| Диаморфин | 1-5 | |
| Декстраморамид | 2-4 | |
| Рацеморфон | 2,5 | |
| Леваметадон | 2 | |
| Метадон | 1,5 | |
| Изометадон | 1-1,3 | |
| Пиминадин | 1 | |
| Проперидин | 1 | |
| Морфин | 1 | |
| Налбуфин | 0,5-0,8 | |
| Слабая | Пиритрамид | 0,7 |
| Гидрокодеин | 0,35 | |
| Пентазоцин | 0,3 | |
| Кодеин | 0,2 | |
| Петидин | 0,1 | |
| Очень слабая | Тимедин | 0,07 |
| Трамадол | 0,05-0,09 |
Промедол
Отечественный синтетический аналог мепередина, примерно в 5-6 раз менее активен, чем морфин, при различных способах введения. Обладает сходной с морфином фармакокинетикой и, соответственно, длительностью болеутоляющего действия, в эквианальгетических дозах отчетливо угнетает дыхание (1, 3, 5). Обычно используется при болевых синдромах средней выраженности в небольших дозах (около 40 мг на 70кг массы тела парентерально), что минимизирует депрессию дыхания и практически нивелирует изменения тонуса гладкомышечных органов (1, 3, 6, 8, 113).
Фентанил
Открытие в период с 1957 по 1962 гг. новых наркотических анальгетиков и среди них фентанила привело к существенному изменению роли опиоидов в анестезиологическом пособии (1, 41, 105). Именно фентанил явился основой таких новых способов обезболивания, как нейролептаналгезия, атаралгезия. В конце 60-х годов фентанил вместе с морфином стал применяться в больших дозах в качестве основного или единственного компонента наркоза.
Фентанил отличается очень высокой болеутоляющей активностью, однако резко угнетает дыхание, особенно у пожилых лиц, вызывает ригидность дыхательной мускулатуры и мышц брюшной стенки (1, 39, 41, 105, 106, 113).
Вводится фентанил преимущественно внутривенно или внутримышечно, при этом скорость развития обезболивающего эффекта составляет 1-3 минуты и 10-15 минут соответственно, а продолжительность аналгезии не превышает 30 минут. Быстрая и выраженная аналгезия обусловлена высокой липоидотропностью фентанила и его способностью проникать через гематоэнцефалический барьер. Основными органами метаболизма являются печень и в значительно меньшей степени почки, в которых осуществляются окислительное дезалкилирование и гидроксилирование фентанила до фенилуксусной кислоты, норфентанила и некоторых других продуктов, которые вместе с небольшой фракцией неизменного фентанила выделяются с мочой (1, 105, 106, 107, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122).
В настоящее время нашли применение ряд аналогов фентанила: альфентанил, суфентанил, ремифентанил, - последний из которых обладает наибольшей продолжительностью действия (1, 41, 105, 106, 107, 114, 123, 124).
Для фентанила и его производных характерно брадикардическое действие обусловленное, по-видимому, активацией центральных парасимпатических механизмов, поскольку брадикардия предупреждается атропином. Другие побочные эффекты выраженные у эталонного анальгетика морфина, при применении фентанила наблюдаются редко (105, 106, 107, 108, 109, 111, 114, 115, 118, 119).
Суфентанил
Синтетический опиоид, в 5-10 раз мощнее фентанила (124). Период полураспределения - 0,72 минут, Т1/2 - 13,7 минут. Почти полностью связывается с белками (92,5%), липофилен. Обладает более быстрым, по сравнению с фентанилом, началом действия (67, 115, 116, 119, 124).
В дозах 10-20 мкг/кг создает надежную антигипертензивную защиту. Не освобождает гистамин (74, 79, 115, 124).
Альфентанил
Синтетический опиоид, в 4 раза слабее фентанила, но обладает более быстрым началом действия и короткой продолжительностью(41, 114, 115).
Альфентанил успешно применяется при коротких операциях. В эксперименте показано, что средние дозы не вызывают изменений центральной гемодинамики, в то время как большие (5 мг/кг) приводят к увеличению ЧСС и СВ (115). Имеются данные, что у некоторых больных после введения альфентанила возникают опасная гипотония, гипертензия или сердечная аритмия ( 110, 112, 115, 119, 121).
Ремифентанил
Первый в этом классе опиоид с очень коротким временем полужизни (менее 10 мин.) за счет высокой степени эстеразной активности метаболизма, что обусловливает быстрое прекращение действия. В дозе 0,4 мкг/кг/мин ремифентанил оказался эффективнее фентанила. Побочные реакции не выявлены (115, 124, 125).
Ремифентанил создает глубокую анальгезию, блокирующую ноцицептивные импульсы (1, 115, 124).
Пентазоцин
Синтетический анальгетик, один из наиболее хорошо изученных представителей нового класса опиоидов, обладающих смешанным агонист - антагонистическим взаимодействием с опиатными рецепторами (1, 126).
Современными методами исследования доказана несостоятельность предположения о том, что особенности фармакологических эффектов пентазоцина обусловлены его селиктивностью к c - опиатным рецепторам. Сравнительно большая избирательность влияния на c - рецепторы характерна для тифлуадома, который хорошо проникает в мозг при парентеральных способах введения (113, 114, 126, 127).
По анальгетической активности пентазоцин в 3-6 раз слабее морфина. В анальгетических дозах вызывает такую же депрессию дыхания, активирует центральные симпатические механизмы, вследствие чего развиваются гипотенезия и тахикардия, может ухудшать коронарный кроваток (67, 68, 113, 114, 115, 127).
В дозах 30-60 мг вызывает анальгезию, соответствующую эффекту морфина в дозе 10 мг. В отличие от морфина пентазоцин может вызывать повышение АД и тахикардию, что связано с активацией адренергических рецепторов (1, 41, 115, 126, 127).
Скорость и длительность болеутоляющего эффекта пентазоцина сходны с таковыми при применении морфина. В связи с низкой биодоступностью (20%) действие пентазоцина при энтеральном применении выражено слабее, чем при внутривенном или внутримышечном способах введения.
Кинетика пентазоцина обычно би-или триэкспоненциальная со средним Т1/2 3-5 ч., плазменным клиренсом 1200-2600 мл/мин и объемом распределения 200-400 литров (114, 115, 126).
Выводится пентазоцин из организма почками, преимущественно в виде метаболитов. Достоинствами его являются слабое проникновение через плаценту и благоприятное влияние на сократительную функцию миометрия, на чем основано его применение в акушерской практике (1, 41, 113, 114, 115, 126).
Бупренорфин
Производное ориповина, обладает высоким аффинитетом к µ - и c- опиатным рецепторам (39, 127, 128). Однако, его фармакологический профиль первично определяется агонизмом к рецепторам и необычно замедленной кинетикой их освобождения. Эти особенности обусловлены наличием в структуре бупренорфина N-бутильной группировки в положении С7, придающей ему сходство с энкефалинами и определяющей его сильное и длительное болеуталяющее действие (39, 74, 76, 128).
Бупренорфин считается первым наркотическим анальгетиком, с помощью которого в определенной степени достигнуто «разделение» болеутоляющих и токсиманических свойств (39). Он обладает очень высокой, близкой к фентанилу, аналгетической активностью и, в отличие от последнего высокой биодоступностью, которая колеблется в зависимости от способов введения от 40-100% (1, 128). При парентеральном введении разовая анальгетическая доза, обеспечивающая достаточный эффект при умеренных и сильно выраженных болевых синдромах, составляет 0,3-0,6 мг на 70 кг массы тела, Т1/2 составляет от 3-5 часов, максимальная анальгетическое действие длится не менее 6 часов (1, 39, 46, 128).
Выраженность угнетающего действия бупренорфина на дыхание совпадает с таковой для морфина в диапазоне оптимальных анальгетических доз, однако, этот нежелательный эффект бупренорфина практически не зависит от дозы (117, 118, 120).
Описано применение бупренорфина без тяжелых последствий в дозе 8мг в сутки в течение нескольких дней подряд. Бупренорфин считается удобным препаратом для терапии послеоперационных болей, причем с этой целью рекомендуется его сублингвальное применение в таблетках (0,2 мг). В этом случае биодоступность бупренорфина составляет в среденм 55%, Т1/2 - 76 мин. при значительной продолжительности действия (117, 118, 119).
Из побочных эффектов отмечают тошноту, рвоту, сонливость, выраженность которых прямо зависит от дозы препарата.
Другие представители этой группы агонист-антагонистов опиатных рецепторов принципиально отличаются от бупренорфина. Они вызывают аналгезию, воздействуя на c - рецепторы, а за счет µ - антоганистической активности препятствует угнетению дыхания(1, 117, 118). К этой группе относятся производные морфина, синтезированные в 80-х годах, - налбуфин, буторфанол, корфанол, пиценадол (1, 116, 117, 118). Все они характеризуются минимальным угнетением дыхания, незначительным влиянием на желудочно-кишечный тракт и сердечно-сосудистую систему. Однако, небольшой диапазон между анальгетическими дозами и дозами, вызывающими психотоматические расстройства, ограничивает их широкое применение в клинике (128).
Налбуфин
Равен морфину по анальгетической активности при внутримышечном введении, при энтеральном приеме эффективность налбуфина в 4-5 раз ниже. Пик концентрации в плазме крови возникает через 30-60 минут, длительность действия 3-6 часов, Т1/2 составляет 2-3 и 7-8 часов при парентеральном и энтеральном введении соответственно (40, 118, 128).
Метаболизируется налбуфин в печени и выделяется с желчью через кишечник. Очень незначительная часть неизмененного налбуфина экскретируется с мочой (74, 118, 129).
Наиболее типичный побочный эффект налбуфина - седативное действие, которое возникает у 36% больных. Другие побочные эффекты бывают редко, например: тошнота, рвота - всего в 6% случаев (40, 128, 129).
Выраженность угнетения дыхания под влиянием налбуфина в дозе 10 мг (внутривенно) сходна с эффектом морфина в такой же дозе. Однако, при увеличении дозы налбуфина депрессия дыхания не усиливается. Налбуфин обладает сравнительно низким психотомиметическим потенциалом, слабым влиянием на моторику желудочно-кишечного тракта, минимальной толерантностью и способностью вызывать физическую зависимость (46, 128, 129).
Буторфанол
По своим свойствам очень близок к налбуфину и отличается от последнего более высокой анальгетической и психотомиметической активностью. Может повышать артериальное давление (105, 130, 131, 132).
Корфанол
Препарат, эффективный при энтеральном применении и обладающий резистентностью к антагонистам опиатов. На основании преимущественно экспериментальных данных предполагают, что корфанол и еще один новый анальгетик - пиценадол - обладают минимальным наркогенным потенциалом (1, 133, 134, 135).
Трамадол
Новый синтетический анальгетик со сравнительно высокой (60-70%) биодоступностью при разных способах введения, быстрым и длительным болеутоляющим эффектом (1, 46). Однако, он уступает морфину по анальгетической активности в 5-10 раз. После внутривенного введения трамадола болеутоляющее действие развивается через 5-10 минут, Т1/2 составляет 6 часов. При энтеральном введении аналгезия возникает через 30-40 минут и не снижается в течение 10 часов(1, 46, 136). В обоих случаях используют трамадол в дозах 100-200 мг на 70 кг массы тела, что обеспечивает создание в крови анальгетической концентрации - 100 нг/мл и более (136, 137).
На фоне трамадола отмечают стабильность параметров кровообращения.
К сожалению, трамадол не лишен характерных для опиодиов не желательных эффектов: часто возникают тошнота и рвота, характерным также считают угнетение дыхания в раннем послеоперационном периоде (137, 138, 139).
Выбор препарата для использования в конкретной клинической ситуации должен определяться в первую очередь фармакокинетическими характеристиками, с обязательным учетом особенностей организма (возраст, состояния систем метаболизма). В ІТаблице 3І приведены сравнительная оценка болеутоляющей активности и длительности действия традиционных и новых наркотических анльгетиков и некоторые показатели из фармакокинетики при энтеральном и парентеральном способах введения (1, 140, 141).
Таблица 3
Сравнительная оценка болеутоляющей активности и длительности действия традиционных наркотических анальгетиков.
| Препарат (международное название) | Путь введения | Эквианальгет-ические дозы (мг) | Анальгезия, ч | Т1/2, ч | |
| Максимум | Длительность | ||||
| Морфин | в/м per os | 10 60 | 0,5-1 1,5-2 | 4-6 4-7 | 2-3,5 |
| Меперилин | в/м per os | 75 300 | 0,5-1 1-2 | 4-5 4-6 | 3-4 |
| Лефорфанол | в/м per os | 2 4 | 0,5-1 1,5-2 | 4-6 4-7 | 12-14 |
| Пентазоцин | в/м per os | 60 180 | 0,5-1 1,5-2 | 4-6 4-7 | 2-3 |
| Налбуфин | в/м | 10 | 0,5-1 | 4-6 | 5 |
| Буторфанол | в/м | 2 | 0,5-1 | 4-6 | 2,5-3,5 |
| Бупренорфин | в/м сублин-гвально | 0,4 8,8 | 0,5-1 2-3 | 6-8 6-12 | 3-5 |
Завершая краткое изложение механизмов действия, влияния на организм наркотических анальгетиков следует подчеркнуть парадоксальность ситуации, которая заключается в том, что даже при наличии определенного набора лекарственных средств, способных эффективно коррегировать болевые синдромы, практически любой выраженности, неудачи в обезболивании могут достигать 70% (134, 135, 136, 139). Одной из причин этого является неправильное применение анальгетиков - неэффективные дозы, нарушение режима и метода введения препаратов. Очень часто не удается достигнуть необходимой концентрации анальгетиков в крови, тем более поддерживать ее длительное время (139, 140).
Создание новых анальгетиков приводит к тому, что у них уменьшается какой-либо побочный эффект, однако, проявляется способность вызывать другие нежелательные реакции (1, 3, 46, 74, 79, 140).
Похожие работы
... кальция в почках ). Витамин В3 - транспорт кальция из кишечника в кость (оссификация кости ). Кальцитонин - поступление кальция из крови в кость. ПРОТИВОАРИТМИЧЕСКИЕ СРЕДСТВА Общая фармакология Поляризация цитоплазматической мембраны зависит от работы калий-натриевого насосов, которые страдают при ишемии - аритмии. Автоматизм Частота может быть изменена при : 1) ...
0 комментариев