Навигация
Для вскрытия чистых металлов, сплавов, сырья различных видов. Заметим, что указанным методом легко вскрывается осмистый иридий;
121066
знаков
12
таблиц
2
изображения
1. для вскрытия чистых металлов, сплавов, сырья различных видов. Заметим, что указанным методом легко вскрывается осмистый иридий;
2. для реактивации и переработки отработанных катализаторов. При обработке дезактивированных катализаторов F2 либо BF3 при t < 100 оС сажа выгорает, а ценный металл переходит в комплексный фторид. В каталитическом процессе происходит его восстановление до металла, и активность катализатора возрастает вновь. В случае переработки отработанных катализаторов обеспечивается одновременное получение платинового концентрата с извлечением ценного компонента > 99% и криолита.
3. для переработки керамических паст: пасты (например, палладий-серебряные) обрабатывают фторирующим агентом, и образовавшийся продукт выщелачивают водой. При этом палладий осаждается в виде гидроксида, а AgF переходит в раствор.
4. в аналитических целях: фторирование BF3 применяется для вскрытия упорных золотосодержащих материалов, содержащих S, Sb, As (реакция идет под слоем фреона).
3. Координационные (комплексные) соединения платиновых металлов
Без преувеличения можно сказать, что химия платиновых металлов есть преимущественно химия координационных соединений. Как типичные переходные элементы, металлы платиновой группы имеют частично заполненные d‑орбитали, вследствие чего характеризуются склонностью к образованию комплексных соединений. При этом они могут иметь разные степени окисления: так, для рутения и осмия известны соединения со всеми возможными степенями окисления центрального атома от 0 до + 8.
Координационными называются соединения, существующие как в кристаллическом состоянии, так и в растворе, особенностью которых является наличие центрального атома (акцептора электронов), окруженного лигандами (донорами электронов). Лиганды способны ступенчато и обратимо отщепляться от центрального атома по гетеролитическому типу. В молекулярном виде координационные соединения могут рассматриваться как состоящие из простых, способных к самостоятельному существованию молекул.
Каждый лиганд в комплексном соединении характеризуется определенным влиянием на другой лиганд, находящийся к нему в транс-положении. Процессы замещения лигандов в комплексах происходят в соответствии с закономерностью транс-влияния И.И. Черняева, которая гласит: «У соединений с квадратным или октаэдрическим строением внутренней сферы, в центре которой находится комплексообразующий атом, скорость реакции замещения всякого атома (или молекулы), связанного с этим центральным атомом, определяется природой заместителя, занимающего противоположный конец диагонали».
Особое место в химии платиновых металлов занимают хлоридные комплексы, наиболее часто встречающиеся в технологии и анализе.
3.1 Хлорокомплексы платиновых металлов
Палладий. Палладий образует хлорокомплексы, в которых проявляет степени окисления +2 и +4. В хлоридных растворах он обычно присутствует в виде хлорокомплексов палладия(II), а также продуктов их акватации и гидролиза. Хлорокомплексы палладия(II) в водных растворах акватируются и гидролизуются легче, чем комплексы других платиновых металлов.
В зависимости от концентрации ионов H+ и Сl- в растворах могут образовываться комплексы состава [Pd(H2O)nCl4-n]n-2, где n может изменяться от 0 до 3 (CPd ~ 10-6 – 10-2 М. В растворах HCl и NaCl с концентрацией Cl- ³ 1 М доминирует форма [PdCl4]2-, а при 0.1 < CCl- < 0.5 М сосуществуют комплексные ионы [PdCl4]2- и [Pd(H2O) Cl3]- Если соотношение Pd(II): Cl – равно 1:1, в растворе доминирует [Pd(H2O) Cl3]-, при отношении 1:10 – в растворе находится смесь комплексов: [Pd(H2O)3Cl]+, [Pd(H2O)2Cl2]0, [Pd(H2O) Cl3]- и [PdCl4]2- Ион [PdCl4]2 – преимущественно присутствует в растворах 1 – 3 М HClO4 или 3 М H2SO4 при концентрации хлорид-иона ~ 0.1 – 1 М.
Предполагается, что анодное растворение палладия постоянным током в растворах НCl сопровождается образованием ионов [Pd(H2O) Cl3]- и [PdCl4]2-]. Легкость, с которой палладий под действием тока переходит в солянокислые растворы, объясняют тем, что выделяющийся на аноде хлор реагирует с палладием и способствует, таки образом, его растворению.
При добавлении щелочи к водным растворам Na2[PdCl4] образуются продукты, представляющие собой полиядерные комплексы, содержащие OH- и H2O – лиганды, либо они являются коллоидными частицами гидроксида Pd(II). В растворах Na2[PdCl4] при рН ³ 3 образуются малорастворимые продукты гидролиза.
Хлорокомплексы палладия(IV) устойчивы только в присутствии сильных окислителей, например, хлора. Стандартный окислительно-восстановительный потенциал системы [PdCl6]2-/[PdCl4]2 – равен 1,29 В.
Платина. Существование хлорокомплексов платины в хлоридных растворах как в степени окисления +2, так и в степени окисления +4 равновероятно. Это обусловлено близостью значений стандартных окислительно-восстановительных потенциалов в системах [PtCl6]2-/[PtCl4]2 – (Eo = +0.726 B) и - [PtCl4]2-/Pt (Eo = +0.78 B).
В зависимости от концентрации хлорид – иона, кислотности среды, температуры в водных растворах могут образовываться акво- и аквогидроксохлоридные комплексы платины(II) состава [Pt(H2O)nCl4-n]n-2, [Pt(H2O)k(OH)m Сl 4m-k]k-2, [Pt(OH)nСl4-n]2-.
В кислых растворах при СCl- > 0,1 М доминирует форма [PtCl4]2- При 10-3 < СCl- < 10-1 М в растворе сосуществуют комплексные ионы [PtCl4]2- и [Pt(H2O) Cl3]-, а при СCl- < 10-3 М – формы [Pt(H2O) Cl3]-, [Pt(H2O)2Cl2]0, [Pt(H2O)3Cl]+.
Действие щелочи на растворы хлорокомплексов платины(II) сопровождается осаждением гидроксида в интервале рН 6–7 (температура oт 18–200С); при кипячении нерастворимые продукты гидролиза образуются при рН ~ 3.
Изучение поведения хлорокомплексов платины(II) в растворах при 150–200 0С в автоклаве показало, что в слабокислых и нейтральных средах возможно протекание реакции диспропорционирования:
[PtCl4]2 ® [PtCl6]2- + Pt0
Платина(IV) образует очень прочные и кинетически инертные комплексы. Общая константа устойчивости [PtCl6]2- – иона оценивается равной lg = 33,9. Несмотря на указанный факт в водных растворах, содержащих комплекс [PtCl6]2-, могут протекать реакции акватации и гидролиза, и в результате образуются различные по составу аквохлоро- и аквогидроксохлорокомплексы Pt(IV): [Pt(H2O)nCl6-n]n-2-, где n = 1; 2; [Pt(H2O)k(OH)mCl6-m-k]k-2, где m = 1; 2; k = 1; 2; [Pt(OH)mCl6-m]2, – где m = 1–6; а также двухъядерные комплексы [Pt2Сl(OH)7 (H2O)2].
На основании анализа опубликованных в литературе данных можно заключить, что гексахлороплатинат(IV) существует в растворах с СHCl > 3 М. При 0.1 < СHCl < 3 М, в 0,5 М растворе KCl, 0,2 и 3 М H2SO4 доминирующим является, наряду с [PtCl6]2-, комплекс [Pt(H2O) Cl5]- При pH = 7–13 преобладают гидроксокомплексы [Pt(OH)5Cl]2- и [Pt(OH)6]2- В щелочных растворах при рН 10–12 реализуются различные варианты реакций замещения с образованием соединений состава транс – [Pt(OH)2Cl4]2-, [Pt(OH)5Cl]2- и [Pt(OH)4Cl2]2- Важно подчеркнуть, что все продукты гидролиза хлорокомплексов платины(IV), даже образующиеся при кипячении в щелочных растворах, хорошо растворимы в воде, и это свойство используется для отделения платины от других платиновых металлов (родия, палладия, иридия).
Родий. Система Rh(III) – H2O – Cl – характеризуется сложными превращениями, включая акватацию, гидролиз, реакции изомеризации и полимеризации. При этом протекающие в данной системе гидролитические процессы не сопровождаются изменением степени окисления центрального атома. Стандартный окислительно-восстановительный потенциал пары [RhCl6]3-/Rh равен +0,43 В, а для пары [RhCl6]2-/ [RhCl6]3 – он составляет +1,2 В.
Образование аквохлорокомплексов родия(III) состава [RhCln(H2O)6-n]3-n, где 0 < n < 6, установлено как при изучении акватации [RhCl6]3- – иона, так и на основе исследований взаимодействия перхлоратных растворов, содержащих гексааквокомплекс [Rh(H2O)6]3+, с HСl. Все аквохлорокомплексы вышеуказанного состава выделены с помощью хроматографических методов и спектрально охарактеризованы.
Если концентрация родия в растворе составляет 5·10-3 – 7.5·10-4 М, гидролиз начинается при рН 3.3–4.5. В щелочных растворах происходит быстрое замещение хлорид-оинов на OH--группы и образование полиядерных гидроксокомплексов с высокой степенью полимеризации.
Явление полимеризации присуще и сильнокислым растворам хлорокомплексов родия(III). Реакции полимеризации протекают с образованием полиядерных макромолекул:
2 [RhCl6]3- Û [Rh2Cl9]3 – +Cl-.
Области доминирования хлорокомплексов родия(III) в HCl – растворах при t = 25 0C с концентрацией по металлу 5´10-4 M приведены в табл. 4.
Таблица 4. Химические формы нахождения комплексов родия (III) в солянокислых растворах| Комплекс | Концентрация HСl, М |
| [RhCl6]3- | 6.0–11.0 |
| [RhCl6]3-, [Rh(H2O) Cl5]2- | 2.0–6.0 |
| [Rh(H2O) Cl5]2-, [Rh(H2O)2Cl4]- | 0.5–2.0 |
| [Rh(H2O)2Cl4]- | 0.25–0.5 |
| [Rh(H2O)2Cl4]-, [Rh(H2O)3Cl3]0 | 0.1–0.25 |
| [Rh(H2O)3Cl3]0 | 0.0–0.1 |
Иридий. В солянокислых и хлоридных водных растворах иридий присутствует в виде комплексов, в которых он проявляет степени окисления +3 и +4. Величина EO([IrCl6]3-/Ir) = 0.77 B,а EO([IrCl6]2-/[IrCl6]3-), определенный разными авторами, имеет значения от +0,87 до +1,02 В. Последний несколько уменьшается с уменьшением ионной силы раствора. На поведение хлорокомплексов Ir(III) и Ir(IV) в растворах существенное влияние оказывает подвижное окислительно-восстановительное равновесие между ними, особенно в слабокислых и слабощелочных растворах.
Предположительно в водных растворах возможно образование аквохлорокомплексов [Ir(H2O)nCl6-n]n-3, гидроксохлорокомплексов [Ir(OH)nCl6-n]3-, биядерных комплексов [Ir2(H2O)mCl7]-, где m = 2; 3; 4; [Ir2(H2O)pCl10]4-, где p = 0; 1; [Ir2(H2O)qCl9]3-, где q = 0; 1; 2; а также полиядерных гидроксохлорокомплексов Ir(III). Однако существует мнение, что димерный комплекс [Ir2Cl9]3 – неустойчив и распадается на ионы [Ir(H2O)2Cl4]-и [Ir(H2O) Cl5]2-.
Хлоридные комплексы иридия(III) менее лабильны, чем хлорокомплексы родия(III). Процессы акватации иона [IrCl6]3 – происходят в ~ 100 раз медленнее, чем иона [RhCl6]3- в аналогичных условиях. Так же как и для Rh(III), скорость реакций акватации уменьшается с уменьшением числа хлорид-ионов во внутренней координационной сфере иридиевого комплекса. Скорость акватации уменьшается при увеличении концентрации катионов щелочных металлов, а при постоянной концентрации изменяется в ряду Li+ > Na+ > K+.
Хлорокомплексы иридия(III) в водных растворах менее устойчивы и более реакционноспособны по сравнению с комплексами иридия(IV).
В результате реакций акватации и гидролиза хлорокомплексов Ir(IV) в зависимости от концентрации H+ и Cl- - ионов, HCl, температуры и времени выдержки в растворах предполагается образование комплексов состава [Ir(H2O)nCl6-n]n-2 и [Ir(OH)nCl6-n]2- Постулируется образование комплексов [Ir(H2O)3Cl3]+, [Ir(H2O)2Cl4], [[Ir(H2O) Cl5]-, [Ir(OH)2Cl4]2-, [Ir(OH)4Cl2]2-, хотя в твердом состоянии ни один из них не выделен.
В солянокислых и хлоридных водных растворах хлорокомплексы иридия(IV) восстанавливаются, причем в качестве восстановителей могут выступать молекулы воды, OH- – ионы (в слабокислых и слабощелочных растворах), а также ионы Сl-:
4 [IrCl6]2- + 2H2O Û 4 [IrCl6]3- + O2 + 4H+
4 [IrCl6]2- + 4OH Û 4 [IrCl6]3- + O2 + 2H2O
2 [IrCl6]2- + 2Cl- Û 2 [IrCl6]3- + Cl2
Указанные реакции являются в той или иной мере обратимыми и протекают без изменения внутренней координационной сферы. Поэтому даже в концентрированных растворах HCl и KCl и в присутствии газообразного хлора в результате восстановления образуются хлорокомплексы иридия(III). Процесс восстановления [IrCl6]2 – ускоряется под действием УФ облучения. Хлорная кислота и перхлорат натрия замедляют его.
Предполагаемые формы существования хлорокомплексов иридия(III) и (IV) даны в табл. 4.
Среди металлов платиновой группы рутений и осмий выделяются многообразием степеней окисления. Ионы этих металлов обладают большим сродством к кислороду, склонностью к образованию оксокомплексов. Поэтому в водных растворах хлорокомплексов рутения и осмия возможно присутствие разнообразных акватированных, гидролизованных полимерных соединений, склонных к окислительно-восстановительным превращениям. Именно в водных растворах хлорокомплексов рутения и осмия можно ожидать существование многих комплексных форм различного заряда.
Таблица 4. Возможные формы нахождения хлорокомплексов иридия(III) и иридия(IV) в водных растворах| Комплекс | Среда | |
| Ir(III) | Ir(IV) | |
| [IrCl6]3- | [IrCl6]2- | >3.0 М HCl |
| [IrH2OCl5]2- [Ir(H2O)2Cl4]- [Ir(OH)2Cl4]3- | [IrCl6]2- [IrH2OCl5]- [Ir(OH)2Cl4]2- | 0,1–3,0 М HCl |
| [Ir(H2O)2Cl4]- | [IrCl6]2- [Ir(H2O) Cl5]- [Ir(OH)2Cl4]2- | 0.01–0.05 М HСl |
| [Ir(OH)2Cl4]2- | pH~7 | |
| [Ir(H2O)4Cl2]+ [Ir(OH)4Cl2]3- | [Ir(H2O)4Cl2]2+ [Ir(OH)4Cl2]2- | pH 7 – 14 |
| Ir2O3·nH2O | IrO2·nH2O | >0.1 М NaOH |
Рутений в хлоридных комплексах находится в степенях окисления +2, +3, +4, +6. В кристаллическом состоянии выделены хлорокомплексы рутения(III): M3[RuCl6]·nH2O, M2[Ru(H2O) Cl5], где M – Na+, K+, Rb+, Cs+, а также димерный комплекс с хлоридными мостиками – K4[Ru2Cl10], хлорокомплексы рутения(IV) типа M2[RuCl6], где M – K+, NH4+, Rb+, Cs+ и биядерная соль K4[Ru2OCl10]·H2O с линейным мостиком Ru – O – Ru. В твердом состоянии выделены оксохлоридные комплексы рутения(VI) состава H2[RuO2Cl4]·3H2O, M2[RuO2Cl4]·nH2O, где M – Rb+, Cs+.
Комплексы рутения в высших степенях окисления способны восстанавливаться в солянокислых растворах хлорид-ионом и кислородом воды, в низших – окисляться кислородом воздуха и протонами. Ориентировочные значения окислительно-восстановительных потенциалов соединений рутения в растворах HCl для следующей схемы превращений составляют:
+0.45 B +0.081 B +0.96 B ([Ru2OCl10]4-) +1.75 B
Ru ® RuCl ® [Ru(H2O) Cl5]2- ® [RuOHCl5]2- ® RuO42 – +1.25 B
à RuO4.
Сведения о хлорокомплексах рутения(II) немногочисленны и противоречивы. Установлено, что они легко окисляются в растворах кислородом воздуха, а при рН = 1.5 – протонами воды. Образование голубых и зеленых соединений неустановленного состава зафиксировано при длительном (в течение 602 суток) выдерживании солянокислых растворов рутения(II) при комнатной температуре.
Хлорокомплексы рутения(III) образуются при взаимодействии RuO4 с растворами HCl (0.5 – 2.0 М) на кипящей водяной бане в течение 30 – 60 мин в присутствии этанола (20 об.%). Гексахлорорутенат(III) – ион – [RuCl6]3 – является доминирующей формой в растворах 6 – 12 М HCl. Однако, даже в концентрированной HCl не исключается существование иона [Ru(H2O) Cl5]2- Эта форма преобладает в 2 – 6 М HCl, а при концентрации HCl < 6 М, помимо моноаквопентахлорокомплекса, в растворе сосуществуют ионы [Ru(H2O)2Cl4]-, [Ru(H2O)3Cl3], цис- и транс – [Ru(H2O)4Cl2]+, [Ru(H2O)5Cl]2+.
Достоверные данные о термодинамической устойчивости хлорокомплексов рутения(III) отсутствуют. Общая константа устойчивости комплекса [RuCl6]3 – оценивается равной lg K = 18.7. Существует мнение, что хлорокомплексы Ru(III) менее термодинамически устойчивы, чем соответствующие комплексы Ru(IV), хотя другие авторы его опровергают. Для различных аквохлорокомплексов Ru(III) известны приближенные константы усточивости, имеются также разрозненные данные по кинетике обмена хлорид-ионов и процессов акватации, константы равновесия между цис- и транс-формами. Комплексы [RuCl6]3- и [Ru(H2O) Cl5]2 – при комнатной температуре акватируются с высокой скоростью, если CHCl < 3M. Однако с уменьшением числа координированных хлорид-ионов константа скорости акватации уменьшается. Так, период полуобмена хлорид-иона на воду увеличивается от нескольких секунд для [RuCl6]3 – до года для [Ru(H2O)5Cl]2+. В среде HСlO4 хлорокомплексы Ru(III) неустойчивы. В диапазоне концентраций HCl 2.8 – 3.9 M при ионной силе, равной 4.0, в присутствии HСlO4 комплекс [Ru(H2O) Cl5]2 – (CRu = 2.5·10-3 моль/л) окисляется до [RuCl6]2-, даже если концентрация кислоты не превышает 5·10-3 М.
Информация о поведении хлорокомплексов рутения(IV) в воде и растворах HCl достаточно противоречива из-за различий в условиях выполнения экспериментов и отсутствия сведений о достижении состояния равновесия в системах. Обычно хлорокомплекс Ru(IV) получают при взаимодействии с соляной кислотой RuO4 или перхлората Ru(IV), а также при сплавлении порошка рутения с хлоратом калия или пероксидом бария с последующей обработкой HCl. В состав хлорокомплексов могут входить акво-, гидроксо- и оксогруппы, а сами соединения могут иметь мономерный и димерный характер.
Комплекс [RuCl6]2 – образуется при восстановлении RuO4 в растворах HCl с концентрацией > 6 М (СRu = 10-3 моль/л, t = 98 oC, t = 100 ч). Предполагается, что в указанных растворах ион [RuCl6]2 – устойчив. В диапазоне концентраций HCl от 1 до 6 М в растворах превалируют две равновесные формы с соотношением Ru: Cl = 1:3 и 1:4. При этом возможно образование комплексов [(RuOH)2(OH)2Cl6]2- и [(RuOH)2Cl8]2-, что не исключает существования и других ионов, например. [Ru(H2O) Cl5]-, [Ru(H2O)2Cl4]o, [Ru(OH)2Cl4]2- В 0.1 М HCl образуются анионные гидроксоформы и катионные комплексы рутения(IV).
В ряде публикаций высказывается предположение, что конечными продуктами гидролиза комплекса [RuCl6]2 – при концентрации HCl менее 5 М являются димерные комплексы [Ru2OCl10]4- и [Ru2O2Cl8]4-, и в растворах с CHCl < 4 M сосуществуют комплексные ионы [Ru2O(H2O)2Cl8]2- и [Ru2O2(H2O)2Cl6]2- Есть мнение, что ион [Ru2OCl10]4 – более устойчив к реакциям акватации и гидролиза по сравнению с ионом [RuCl6]2- При концентрации HCl от 6 до 11 М в растворе, вероятно, доминирует форма [Ru2OCl10]4-, хотя другие авторы считают, что даже в 11 М HCl в растворе возможно существование продуктов ее акватации: [Ru2O(H2O) Cl9]3- и [Ru2O(H2O)2C8]2- В солянокислых растворах, содержащих ион [Ru2OCl10]4 – (CRu = 10-4 моль/л, t = 25 oC, CHCl = 2–6 M) равновесие устанавливается годами.
Комплекс [Ru2OCl10]4 – присутствует в растворах 3.4 М HCl и 3.8 М LiCl. Комплекс [Ru2O(H2O)2Cl8]2 – доминирует в растворах 1.7 – 3.4 М HCl, 2.5 – 3.8 М LiCl, 3.0 – 4.5 M NaCl. В растворах «бурой» соли рутения (CRu = 4.85·10-5 моль/л), содержащих 1 М HCl и 1 М HСlO4, в зависимости от концентрации хлорид-иона образуются комплексы с соотношением Ru: Cl = 1:1 и 1:2, которым приписывают состав {[Ru2(OH)4Cl2]2+}m и [Ru2(OH)4Cl4]o.
Предложена схема акватации и гидролиза солей K2[RuCl6] и K4[Ru2OCl10] с образованием биядерных частиц, согласно которой при концентрации HCl от 0.1 до 6.0 М в растворах доминируют биядерные (а, возможно, и полиядерные) формы рутения(IV), содержащие группировки с гидроксомостиками
H
|
![]()
![]() O
O
Ru Ru
O
|
H
Образование иона [(RuOH)2Cl8]2 – предполагается при CHCl > 5 М, а иона [(RuOH)2(OH)2Cl6]2- в интервале концентраций 1 – 5 М HCl. Не исключается существование в растворах и других мономерных и полимерных аквогидроксохлорокомплексов Ru(IV). Важно отметить, что в солянокислых растворах [RuCl6]2- и [Ru2OCl10]4 – зафиксированы идентичные продукты гидролиза. Обратный переход в высшие хлорокомплексы идет только в сторону образования комплекса [Ru2OCl10]4-.
Спектрофотометрическим методом изучено комплексообразование в системах [RuO(H2O)4]2+ - HCl (LiCl, NaCl) – HСlO4 и показано, что время достижения равновесия в растворах изменяется от 4 ч (в 10 М HCl) до 12 мес. (в 0.1 М NaCl), а в растворах с CCl- < 0,1 моль. л состояние равновесия при 20 оС не устанавливается в течение трех лет. При замене катиона фонового электролита в ряду Na+ – Li+ – H+ равновесие реакций образования хлорокомплексов рутения(IV) смещается в сторону более координационно насыщенных химических форм. Более корректно, однако, мнение, что исходной формой в растворах 1 – 4 М HСlO4 является не мономерный комплекс [RuO(H2O)4]2+, а тетрамер [Ru4(OH)4]4+. При увеличении рН таких растворов увеличивается доля нейтральных форм, начинается поликонденсация и формирование осадка RuO2·nH2O. Введение Сl- – ионов в раствор вызывает разрушение полимера, внедрение Cl- – ионов во внутреннюю координационную сферу рутения с образованием нейтральных и анионных аквогидроксохлорокомплексов.
Результаты исследований «старения» растворов [Ru2OCl10]4- в 0.1 М HCl позволили сделать вывод, что с увеличением времени выдерживания растворов происходит образование катионных комплексов полимерного строения, возможно, вида {[(RuOH)2(H2O)4(OH)2]4+}. Если к таким растворам добавлять соляную кислоту, появляется фиолетовое окрашивание, однако в процессе гидролиза [Ru2OCl10]4 – «фиолетовая» форма не образуется. При добавлении соляной кислоты к гидроксиду рутения(IV) также наблюдается появление фиолетовой окраски, по-видимому, за счет образования соединения с соотношением Ru: Cl = 1:2.
Хлоридные комплексы рутения(IV) можно получить и окислением соединений с более низкой степенью окисления. Возможные продукты окисления аквохлорокомплексов Ru(III) в среде HCl – HClO4 в зависимости от концентрации HCl представлены в табл.
Хлорокомплексы рутения(IV) в солянокислых и хлоридных водных растворах подвергаются не только реакциям акватации, гидролиза, полимеризации, но и восстановлению до соединений рутения(III). Устойчивость к восстановлению определяется составом раствора, концентрацией комплекса и температурой. Так, например, в присутствии NaCl восстановление рутения(IV) в форме [RuCl6]2 – не происходит даже при кипячении раствора. Концентрированные по металлу растворы хлорокомплексов рутения(IV) также весьма устойчивы к восстановлению.
Таблица. Состав продуктов окисления аквохлоридных комплексов Ru(III) в среде HCl – HClO4 (m = 4.0)| Состав комплексных форм в растворе | HCl, М |
| [Ru2O2(H2O)2Cl6]2-- | 0.1 |
| [Ru2O2(H2O)2Cl6]2-, [Ru2O(H2O)2Cl8]2-, | 0.1 – 1.2 |
| [Ru2O2(H2O)2Cl6]2-, [Ru2O(H2O)2Cl8]2-, [RuCl6]2- | 1.2 – 2.8 |
| [RuCl6]2- | 2.8 – 3.9 |
Предполагается, что восстановлению биядерного комплекса предшествует его превращение в мономер по реакции
Ru2O6+ + 2H+ à 2Ru4+ +4H2O
Eo ([Ru2OCl10]4-/[Ru(H2O) Cl5]2-) равен 0.96 В. При уменьшении концентрации Cl- – ионов и особенно H+ – ионов потенциал пары Ru(IV)/Ru(III) увеличивается, а потенциал кислорода уменьшается и, следовательно, условия существования рутения(III) становятся термодинамически более благоприятными.
Тенденция к деполимеризации комплекса [Ru2OCl10]4 – увеличивается с ростом концентрации HCl и уменьшается с увеличением концентрации рутения. В концентрированной HCl при 20 оС восстановление [Ru2OCl10]4 – до Ru(III) сопровождается полной деполимеризацией по реакции
[Ru2OCl10]4 – +2H+ + 4Cl- à 2 [RuCl6]3- + Cl2 + H2O
через 7 дней, если CRu = 6 мг/л, и через 60 дней, если CRu = 18 мг/л. Наиболее вероятно, что деполимеризация происходит одновременно с восстановлением и нагревание до 60 – 80 оС не изменяет характера процессов восстановления и деполимеризации, а только ускоряет их.
Оксохлорокомплексы рутения(VI) получают при взаимодействии хлора и соляной кислоты (или насыщенных солянокислых растворов RbCl и CsCl) c тетраоксидом рутения. Хлорокомплекс(VI) – [RuO2Cl4]2 – устойчив только в присутствии сильных окислителей и сам обладает окислительными свойствами. Спектрофотометрическим и экстракционным методами показано, что ион [RuO2Cl4]2 – доминирует в интервале концентраций 0.5 – 3 М HCl; ему сопутствует комплекс [RuO2(H2O)2Cl2]. В отсутствии окислителей комплексы Ru(VI) восстанавливаются до соединений Ru(IV) мономерного или полимерного характера в зависимости от концентрации HCl в растворе. При этом чем выше концентрация кислоты, тем быстрее идет процесс восстановления. В водных растворах хлорокомплексы Ru(VI) не устойчивы и диспропорционируют с образованием RuO2 и RuO4
Таким образом, в хлоридных и солянокислых водных растворах наиболее вероятно нахождение рутения в степенях окисления +3 и +4.
Хлоридные комплексы осмия исследованы меньше, чем хлорокомплексы других платиновых металлов. Хлорокомплексы осмия известны в степенях окисления +2, +3, +4, +6, но низкие степени окисления (+2, +3) для осмия менее характерны, чем для рутения.
В кристаллическом состоянии выделены хлорокомплексы осмия(III) – M3[OsCl6]·nH2O, где M – Na+, K+, NH4+, осмия(IV) типа M2[OsCl6], где M – K+, NH4+, Cs+, Ag+, а также биядерные оксохлорокомплексы, аналоги «бурой» соли рутения(IV) – M4[Os2OCl10], где M – K+, Cs+, NH4+. Все биядерные оксокомплексы в твердом состоянии диамагнитны, их строение подтверждено рентгеноструктурными исследованиями. В твердом состоянии выделены оксохлоридные диамагнитные комплексы осмия(VI) состава M2[OsO2Cl4], где M – K+, Cs+, NH4+, содержащие линейную группировку O = Os = O.
Имеются указания, что в спиртовых растворах комплексa [OsCl6]3 – образуется сине-фиолетовое соединение осмия(II) – [OsCl6]4- Комплексный ион [OsCl6]3 – можно получить при длительном нагревании [OsCl6]2- c HCl. В водных растворах хлорокомплексы осмия(III) неустойчивы и разлагаются с выделением гидратированного оксида Os2O3·nH2O. Комплексный ион [OsCl6]3- в разбавленной HCl также гидролизуется, причем равновесие в растворах с COs = 0.01 – 0.05 моль/л при комнатной температуре достигается в течение нескольких недель.
Термодинамические характеристики для хлорокомплексов осмия(III) неизвестны.
Наиболее устойчивыми и относительно хорошо изученными являются хлорокомплексы Os(IV) – [OsCl6]2- Чаще всего эти соединения получают взаимодействием OsO4 с концентрированной соляной кислотой при нагревании, иногда в присутствии восстановителей. Стандартный окислительно-восстановительный потенциал Eo системы [OsCl6]2-/[OsCl6]3 – равен 0.85 В.
В кинетическом отношении [OsCl6]2 – наиболее инертен по сравнению с аналогичными хлорокомплексами других платиновых металлов. Акватация [OsCl6]2 – при комнатной температуре происходит медленно и лигандный обмен незначителен.
Константа скорости акватации (k65) при 80 оС (m = 0.5 – 1.32) составляет 3.3·10-6 с-1, а реакции анации иона [Os(H2O) Cl5] – (k56) в 3.3 – 3.8 М HCl равна 2·10-5 М-1с-1. Константа скорости обмена хлорид-ионов в 8.8 М HCl при 80 – 100 оС составляет величину 3.1·10-6 с-1 Хлорокомплексы осмия(IV) легко разлагаются в растворах под действием света с выделением осадка OsO2·nH2O. Превращения под действием света характерны для комплексов всех платиновых металлов, но в случае комплексов осмия они имеют особенно большую скорость. В растворах [OsCl6]2- в HCl во времени образуются аквохлорокомплексы состава [Os(H2O) Cl5]-, [Os(H2O)2Cl4]o, [Os2O(H2O)2Cl8]2-.
При хранении растворов K2[OsCl6] (COs = 5·10-5 моль/л) в HCl в темноте (t = 20 – 25 oС) последние устойчивы в интервале концентраций HCl от 0.01 до 11.04 М в течение 6–7 месяцев, а доминирующей формой существования осмия в них является комплекс [OsCl6]2- Под действием рассеянного солнечного света реакция акватации иона [OsCl6]2 – наблюдается даже в 10 М НСl. В этих условиях в растворах 3.3 – 10.9 М HCl сосуществуют комплексные ионы [OsCl6]2- и [Os(H2O) Cl5]- Последний является доминирующей формой в растворах HCl с концентрацией 1.14 – 3.36 М. В растворах 1.14 М HCl обнаруживаются комплексы [Os(H2O) Cl5]- и [Os(H2O)2Cl4]o. Если CHCl < 0.12 М, то при нагревании и на свету образуется OsO2 ·xH2O. При нагревании [OsCl6]2- в воде при 100 оС в течение 1 ч все хлорид-ионы «уходят» из внутренней сферы комплекса, но состав продуктов гидролиза в этом случае не исследовался.
Наиболее детально изучены продукты акватации и гидролиза, образующиеся в растворах K2[OsCl6] в 1 – 3 М H2SO4. Хроматографией на колонке с диэтиламиноэтилцеллюлозой выделены и спектрально охарактеризованы следующие хлорокомплексы: [Os(H2O) Cl5]-, цис – [Os(H2O) OHCl4]2-, фац – [Os(H2O) (OH)2Cl3]-, мер – [Os(H2O)2(OH) Cl3]-, димер с оксомостиком фац – {[(H2O) (OH) – Cl3Os]2m-O}2- и димеры с мостиковой OH‑группой фац – {[(H2O) (OH) – Cl3Os] (m-OH)}-, транс – {[(H2O)2(OH) Cl2Os]2(m-OH)}+ и цис – {[(H2O)2(HO) – Cl2Os]2(m-OH)}. Такие же комплексы образуются при осторожном восстановлении OsO4 сульфатом железа(II) в растворах HCl. Хлорной кислотой комплексы Os(IV) окисляются до OsO4.
Биядерные оксохлорокомплексы Os(IV) – [Os2ОCl10]4- в водных растворах менее устойчивы по сравнению с комплексом рутения(IV) – [Ru2OCl10]4- Связь Os – O – Os отличается меньшей прочностью, поэтому в водных растворах она легко разрушается, и продуктами гидролиза оказываются мономерные комплексы состава [Os(OH) Cl5]2-, [Os(OH)2Cl4]2-, [Os(OH)3Cl3]2-, фац- и мер – [Os(H2O) (OH)2Cl3]- и [Os(H2O)2(OH) Cl3]o.
Хлорокомплексы осмия(VI) – [OsO2Cl4]2- – получают взаимодействием OsO4 с солянокислыми растворами KCl, NH4Cl или CsCl, либо действием HCl на K2[OsO2(OH)4].
Осмий(VI) обладает особенно высоким сродством к кислороду. В отличие от [RuO2Cl4]2 – оксохлорокомплекс Os(VI) – [OsO2Cl4]2- в растворах более устойчив.
3.2 Сульфатокомплексы платиновых металловСульфатокомплексы платиновых металлов образуются в процессах переработки медно-никелевых шламов сульфатизацией и имеют чрезвычайно сложное строение. Характерной особенностью сульфатокомплексов является их многоядерность. Ионы платиновых металлов в составе сульфатокомплексов провляют различные степени окисления одновременно. В качестве примера можно рассмотреть сульфатокомплексы иридия.
В течение 50 лет французский химик М. Делепин занимался исследованиями сульфатокомплексов иридия. В 1906 г. он впервые сообщил о химическом соединении темно-зеленого цвета, полученном при кипячении раствора гексахлороиридата(III) Na3[IrCl6] и (NH4)2SO4 в конц. серной кислоте, которое он первоначально считал простым комплексом иридия(III) и приписывал ему формулу K4H2NIr3(SO4)6·3H2O. Дальнейшее изучение данной соли показало, что ей отвечает состав K4[Ir3N(SO4)6(H2O)3], причем иридий имеет степень окисления (III, IV, IV) [17]. Если проводить синтез в отсутствие ионов NH4+, то взамен соли Делепина получится сине-зеленая соль Лекок де Буабодрана K10[Ir3O(SO4)9]·3H2O, являющаяся кислородным аналогом соли Делепина и, соответственно, содержащая два атома Ir(III) и один Ir(IV).
Взаимодействие хлорокомплексов иридия с серной кислотой в водном растворе протекает через образование промежуточных продуктов гидролиза хлоридов типа IrxOyClz, которые выделяются в твердую фазу. В серной кислоте при температуре, близкой к ее температуре кипения и примерно равной 300°С, эти соединения постепенно растворяются с образованием комплексных сульфатов [Ir3O(SO4)9]10- Независимо от того, в какой степени окисления находился иридий в исходном комплексе, равновесными формами оказываются термодинамически наиболее устойчивые сульфаты со смешанной степенью окисления центрального атома. Обнаружено, что если исходить из гексахлороиридата(IV), то образование сульфатокомплексов протекает с выделением стехиометрического количества элементного хлора, следовательно, в качестве восстановителя выступает Сl-–ион. Если же в реакцию с H2SO4 вступает гексахлороиридат(III), то выделяется SO2, то есть окислителем является сама серная кислота:
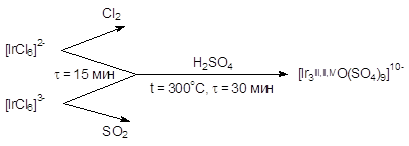 (1)
(1)
Наибольший интерес для оценки химического состояния иридия в процессах жидкостной сульфатизации представляет система Ir(OH)4 – конц. H2SO4, так как гидроксид иридия(IV) является одной из вероятных форм нахождения иридия в анодных медно-никелевых шламах.
Взаимодействие Ir(OH)4·xH2O (сIr > 1·10-3 М) с серной кислотой можно представить протекающим по следующей схеме:
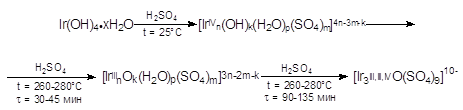 (2)
(2)
Установлено, что в начальный момент реакции гидроксида иридия(IV) с конц. H2SO4 при нормальных условиях (поскольку данный процесс экзотермичен, то нет необходимости повышать температуру) образуется «синий» сульфат иридия(IV), который сохраняет полимерную структуру исходного гидроксида. Через 30–45 мин нагревания при температуре 260–280°С наблюдается появление следующей формы: она оптически прозрачна в видимой и слабо поглощает в УФ–области, что характерно для сульфатов иридия(III) вероятного состава [Ir3(SO4)11H2O]13- Так называемый «бесцветный» сульфат Ir (III, III, III) выделен в виде Cs11H2[Ir3(SO4)11H2O]. Дальнейшее нагревание сопровождается частичным окислением Ir(III) до Ir(IV) конц. серной кислотой, более глубокой гидратацией и деполимеризацией с образованием равновесного «зеленого» оксосульфата иридия (III, III, IV) – [Ir3O(SO4)9]10- При введении ионов аммония в систему Ir(OH)4·xH2O – конц. H2SO4 образуется устойчивый m-нитридосульфат иридия [27].
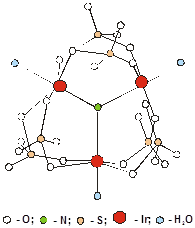
Рис. 1. Структура комплекса [Ir3(m3-N) (SO4)6(H2O)3]4-
Первое рентгеноструктурное исследование сульфатного комплекса платинового металла, выполненное в 1971 г., показало, что соединение K4[(Ir3(m3-N) (m-SO4)6(H2O)3] имеет трехъядерное строение [28]. Центральный атом азота, лежащий на тройной оси, координируется с тремя атомами иридия, сохраняющими октаэдрическую точечную симметрию и соединенными попарно двумя сульфатогруппами. Структура комплекса изображена на рис. 1. Октаэдрическая координация вокруг атома иридия дополнена молекулами воды в транс-положении к атому азота. Расположение атомов азота, иридия и молекул воды – фактически плоскостное (максимальное отклонение 0.01Å).
Структура однотипного оксосульфата неизвестна. Можно говорить лишь о том, что оно аналогично выше приведенному.
Оксосульфатокомплекс Ir (III, III, IV) в водных растворах серной кислоты подвергается ступенчатому гидролизу, первая стадия которого протекает медленно, без разрушения трехъядерной структуры комплексного аниона [Ir3III,III,IVO(SO4)9]10- и сопровождается замещением концевых сульфатогрупп молекулами воды:
[Ir3III,III,IVO(SO4)9]10- + 3H2O ![]() (Ir3III,III,IVO) (SO4)6(H2O)3]4- + 3SO42-
(Ir3III,III,IVO) (SO4)6(H2O)3]4- + 3SO42-
В состав амминокомплексов входит в качестве лиганда молекула аммиака. Амминокомплексы образуются в процесах аффинажа, например, плоскоквдратный комплекс состава [Pd(NH3)2Cl2]. Комплекс транс-строения характеризуются крайне низкой растворимостью в воде, благодаря чему используется для количественного выделения палладия из растворов аффинажного производства (см. раздел 5).
3.4 Нитрокомплексы платиновых металлов
Образуются при нитровании путем замещения лигандов во внутренней сфере комплексов на ион NO2- В нитрокомплексах платиновые металлы проявляют более низкие характерные для них степени окисления. Известны следующие комплексы: [M(NO2)4]2-, где M = Pd(II), Pt(II); [M¢(NO2)6]3-, где M¢ = Rh(III), Ir(III); [M¢¢NO(NO2)4OH]2-, где M¢¢ = Ru, Os. Различие в составе и свойствах указанных соединений обусловливает возможность разделения близких по свойствам платиновых металлов. Они хорошо растворимы в водных растворах и не гидролизуются. Подробно поведение нитрокомплексов рассмотрено ниже.
4. Технологические аспекты аффинажа платиновых металлов 4.1 «Классическая» технология
Аффинаж (от французского «affiner» – очищать) – заключительная стадия переработки различных видов платиносодержащего сырья. Конечными продуктами аффинажа являются платиновые металлы в виде порошков и слитков, которые по чистоте должны отвечать требованиям ГОСТ.
На аффинаж поступают первичное сырье (концентраты, образующиеся при переработке шламов, «шлиховая платина») и вторичное сырье (электронный лом, дезактивированные катализаторы, отработанные электролиты и др.). Выбор конкретной технологической схемы и оптимальных режимов технологических операций зависит от количественного и качественного состава продуктов, подлежащих переработке. Чтобы добиться максимального извлечения ценных металлов с минимальными потерями, а также с целью сокращения незавершенного производства, необходимо рационально сочетать в технологическом цикле известные методы и приемы, которые будут изложены ниже. Варианты технологических схем переработки некоторых видов сырья даны в Приложении.
Первоначально на примере «шлиховой платины» рассмотрим аффинаж по классической схеме, предусматривающей выделение платиновых металлов в виде трудно растворимых комплексных соединений с последующим их прокаливанием и получением аффинированных порошков. Следует подчеркнуть, что в настоящее время практически повсеместно и в нашей стране, и за рубежом для вскрытия платиносодержащего сырья используется процесс гидрохлорирования, в результате которого все металлы платиновой группы, а также золото переходят в раствор в виде комплексных хлоридов (серебро остается в твердом остатке). Важно здесь обратить внимание на поведение осмия в процессе гидрохлорирования. Установлено, что в растворах гидрохлорирования металлического осмия и осмийсодержащих продуктов (температура процесса 80 оС, расход хлора – 1 л/час, СHCl = 0.5 – 6 М) присутствует Os(VIII) в виде OsO4. С ростом концентрации HCl в растворе, содержащем OsO4, его переход в газовую фазу растет. Отсюда вытекает принципиальный вывод о необходимости улавливания тетраоксида осмия на стадии вскрытия сырья во избежание потерь этого весьма редкого и дорогого платинового металла. Аналогичным образом ведет себя осмий и при «царсководочном» вскрытии. Растворение «шлиховой платины» в «царской водке» – традиционный, хотя и несколько устаревший метод растворения продуктов, содержащих платину. Остановимся подробнее на поведении платиновых металлов при растворении «шлиховой платины» и их последующем разделении и выделении.
4.1.1 Аффинаж платиныРастворение «шлиховой платины» в «царской водке» осуществляется при температуре 70–85 оС, причем сырье загружается в предварительно нагретую до указанной температуры соляную кислоту, а затем добавляется рассчитанное количество HNO3. Внешними признаками конца растворения является бурное вспенивание и выделение паров оксидов азота по реакциям:
3HCl + HNO3 = NOCl + Cl2 + 2H2O
NOCl ® NO + Cl
2NO + O2 ® NO2.
Таким образом, не исключено выделение весьма реакционноспособного атомарного хлора, взаимодействующего с благородными и неблагородными металлами. Последние переходят в хлориды (FeCl3, CuCl2 и др.), а платина, палладий и золото – в хлоридные комплексы в соответствии с реакциями:
3Pt + 18HCl + 4HNO3 = 3H2[PtCl6] + 4NO + 8H2O
3Pd + 18HCl + 4HNO3 = 3H2[PdCl6] + 4NO2 + 8H2O
Au + 4HCl + HNO3 = H[AuCl4] + NO + 2H2O.
Палладий даже при растворении в «царской водке» частично переходит в тетрахлоропалладиевую кислоту H2[PdCl4], где проявляет типичную для него степень окисления +2. Некоторое количество платины и иридия образуют соединения H2[PtCl4] и H2[IrCl6], соответственно. Небольшая часть платины может образовывать гексахлороплатинат(IV) железа по реакции:
3H2[PtCl6] + 2FeCl3 = Fe2[PtCl6]3 + 6HCl.
В присутствии азотной кислоты также образуются нитрозохлоридные соединения платиновых металлов, которые выпадают в виде желтого осадка. Образование нитрозохлоридных соединений протекает по реакциям:
H2[PdCl4] + 2NOCl = (NO)2[PdCl4] + 2HCl
H2[PtCl6] + 2NOCl = (NO)2[PtCl6] + 2HCl
H2[IrCl6] + 2NOCl = (NO)2[IrCl6] + 2HCl
Если нитрозохлоридные соединения не разрушать, то они будут оставаться в нерастворимом остатке. Разрушение их осуществляется нагреванием раствора с добавлением воды при температуре 105–110 оС в результате протекания следующих реакций:
(NO)2[PtCl6] + H2О = H2[PtCl6] + NO+ NO2.
Аналогично идут реакции для палладия и иридия. Конец разрушения определяется по прекращению выделения бурых паров оксидов азота и вскипания.
Для растворения «царской водкой» обычно применяются 33%-ная соляная и 65%-ная азотная кислоты. Расход кислот на 100 кг шлиховой платины составляет: HCl – 500 л, HNO3 – 100 л. В результате растворения «шлиховой платины» в «царской водке» в раствор переходит большая часть (90% при первом растворении) платины, почти весь палладий, около 60% родия, 15% иридия, 90% золота и 100% железа и меди. Часть иридия, родия, платины и серебро в виде AgCl остаются в нерастворившемся остатке. Процентное извлечение платиновых металлов при растворении в «царской водке» определяется составом конкретной партии перерабатываемой «шлиховой платины». В зависимости от этого выход первого нерастворимого остатка изменяется от 10 до 15%.
Растворы, полученные после вскрытия в «царской водке», обрабатывают HCl с целью удаления остатков азотной кислоты. Эта операция проводится при постоянной температуре 120–125 оС. После упаривания раствор охлаждается и отфильтровывается от первого нерастворимого остатка, который еще содержит некоторое количество платины. Поэтому, собрав достаточное количество нерастворимого остатка, производят повторное его растворение примерно по тому же режиму, что и основную «шлиховую платину». Полученный второй нерастворимый остаток содержит мало платины и богат редкими платиновыми металлами, на извлечение которых он направляется.
Растворы после растворения «шлиховой платины» и первого нерастворимого остатка и упаривания с соляной кислотой поступают на доводку, цель которой как можно полнее перевести Pd(IV) в Pd(II), Ir(IV) в Ir(III), Au(III) в золото элементарное. Необходимость доводки определяется тем, что последующая операция – это выделение (осаждение) платины в виде гексахлороплатината(IV) аммония (NH4)2[PtCl6] (ХПА), а Ir(IV) и Pd(IV) также образуют однотипные нерастворимые соли. В то же время (NH4)2[PdCl4] и (NH4)3[IrCl6] хорошо растворимы в воде. Необходимо обратить внимание на то, что в процессе доводки как можно большая часть платины должна остаться в степени окисления+4, чтобы обеспечить высокий выход первого гексахлороплатината(IV) аммония (комплексная соль (NH4)2[PtCl4] подобно (NH4)2[PdCl4] хорошо растворима). Родий в процессе доводки степень окисления не меняет. Доводка осуществляется так: раствор при температуре 120–125 оС упаривается до плотности 1.38, затем обрабатывается 15%-ным этиловым спиртом, расход которого – 250–300 мл на 200 кг шлиховой платины. Этиловый спирт расходуется на процесс восстановления по следующим реакциям:
12H2[IrCl6] + C2H5OH + 3H2O = 12H3[IrCl6] + 2CO2
6H2[PdCl6] + C2H5OH + 3H2O = 6H2[PdCl4] + 2CO2+ 12HCl
4H[AuCl4] + C2H5OH + 3H2O = 4Au + 2CO2+ 16HCl.
Этиловый спирт разрушает и возможно присутствующие в растворе остатки азотной кислоты.
Обрабатываемый раствор прогревается в течение 30 мин и опробуется на качество ХПА. Конец спиртования определяется по следующим признакам:
· по прекращению вспенивания и выделения газообразных оксидов;
· по запаху спирта;
· по пробе на качество (NH4)2[PtCl6]: если окраска ХПА не характерна, то добавляют еще некоторый избыток этилового спирта и прогревают реакционную смесь.
Раствор разбавляют водой до плотности 1.16 – 1.18 и осаждают золото, для чего в раствор вводится щавелевая кислота H2C2O4 расчета 5 г на 1 кг «шлиховой платины», и содержимое котла прогревается в течение 3‑х часов при температуре 95–100 оС. В случае избытка H2C2O4 восстанавливается не только золото, частично иридий, палладий, но и платина по реакции:
H2[PtCl6] + H2C2O4 = Pt + 2HCl + 2CO2
Раствор после нагревания с H2C2O4 вновь разбавляют до плотности 1.16–1.18 и обрабатывают 20%-ным раствором сахара (расход сахара – 2–3 г. на 1 кг шлиховой платины), прогревают в течение 50–60 мин при температуре 95–100 оС и проверяют полноту осаждения золота. Раствор после фильтрации от осадка золота поступает на осаждение ХПА, а золотосодержащий кек – на извлечение золота.
Для получения крупного кристаллического осадка ХПА раствор должен иметь плотность 1.16. Получение крупнокристаллического осадка обеспечивает лучшее качество ХПА, т. к. мелкокристаллическийо осадок имеет более развитую поверхность и сорбирует на себе различные примеси. С целью получения крупнокристаллического осадка ХПА применяют растворы хлорида аммония различной концентрации: сначала вводят 5%-ный раствор NH4Cl, затем 12,5%-ный и, наконец, 25%-ный. По данным практики, количество хлорида аммония, вводимого с растворами различной концентрации, должно быть следующим:
· 5%-ный раствор составляет 20% от общего расчетного количества NH4Cl;
· 12,5%-ный – 20% от общего количества NH4Cl;
· 25%-ный – 50% от общего количества NH4Cl.
Осаждение ведется на холоду по реакции:
Н2[PtCl6] + 2NH4Cl = (NH4)2[PtCl6] + 2HCl.
Гексахлороплатинат(IV) железа также взаимодействует с NH4Cl с образованием ХПА:
Fe2[(PtCl6)]3 + 6NH4Cl → 3 (NH4)2[PtCl6] + 2FeСl3.
После проверки на полноту осаждения раствор с осадком переносится на фильтр и фильтруется. Желательно очень быстро отделить раствор от осадка, в том числе и потому, что при продолжительном соприкосновении раствора с осадком последний очень плохо отмывается от примесей и, в особенности, от солей железа и палладия. Несмотря на эти меры предосторожности, ХПА сорбирует небольшое количество солей, поэтому необходимо проводить промывку осадка, которая производится 5%-ным раствором хлорида аммония, причем о полноте отмывки от железа судят по реакции с тиоцианатом аммония:
FeСl3 + 3NH4CNS = Fe(CNS)3 + 3NH4Cl.
О полноте отмывки от палладия судят по реакции с диметилглиоксимом, при этом палладий с диметилглиоксимом образует внутрикомплексное соединение [(CH3)2C2N2O2H]2Pd. Отмытый ХПА подсушивается и направляется на прокаливание.
Получение губчатой платины производится в электрических печах сопротивления. ХПА начинает разрушаться при температуре 300 оС по реакции:
3 (NH4)2[PtCl6] → 3Pt + 2NH4Cl + 16HCl + 2N2.
Во избежание большого пылеуноса температура должна подниматься по строго разработанному режиму. Операция длится 10 часов. К концу операции температура поднимается до 1100 оС. Полученная платиновая губка не должна содержать непрокаленный ХПА. Цвет губки – светло-серый, и при ударе она должна мяться, не рассыпаясь в порошок. Если губка отвечает кондиции, то поступает на плавку, но большей частью она не отвечает требованию ТУ и направляется на переаффинаж – растворение в «царской водке» и все последующие операции аффинажа. Платиновая губка 2, как правило, отвечает требованиям кондиции.
Растворы и промывные воды после осаждения I‑го ХПА направляются на упаривание с целью концентрирования солей в растворе, дополнительного выделения гексахлороплатината, а также осаждения солей иридия(IV) и палладия(IV). Это достигается добавлением азотной кислоты, при этом обеспечивается переход Ir(III) → Ir(IV); Pd(II)→ Pd(IV); остатки Pt(II)→ Pt(IV). Поскольку в растворе имеется избыток NH4Cl, то выпадают соответствующие соли, а именно: (NH4)2[PdCl6], (NH4)2[PtCl6], (NH4)2[IrCl6]. Раствор отфильтровывают от осадка солей, и осадок прокаливается. После прокаливания получается губка так называемой «иридистой» платины, которая содержит в среднем платины – 50–55%, иридия до 30%, палладия 17–18%, родия – 0,05 – 0,07%. В этом продукте еще много платины и поэтому производят переаффинаж с целью ее выделения. Переаффинаж производится по схеме основного аффинажа с небольшим изменением, которое касается в первую очередь дозировки различных реагентов.
Нерастворимый остаток от растворения в «царской водке» «иридистой» платины содержит большое количество иридия и родия и направляется на их извлечение, а растворы после осаждения ХПА содержат много палладия и поступают на аффинаж палладия.
4.1.2 Аффинаж палладия
Палладиевый раствор упаривается в котлах при температуре 110–120 оС. В раствор постепенно вводится аммиак. Перед введением аммиака палладий в растворе находится в виде тетрахлоропалладата(II) аммония (NH4)2[PdCl4]. В том случае, если аммиак вводится в избытке, то должна протекать конечная реакция:
(NH4)2[PdCl4]+ 4NH3 = [Pd(NH3)4] Cl2 + 2NH4Cl.
В действительности аммиак прибавляется постепенно, и поэтому первоначально часть палладия переходит в тетраамминпалладий(II), а другая часть палладия – остается в форме тетрахлоропалладат(II) – иона. Эти комплексы взаимодействуют друг с другом с образованием нерастворимой соли Вокелена состава [Pd(NH3)4] [PdCl4] по реакции:
(NH4)2[PdCl4] + [Pd(NH3)4] Cl2 = [Pd(NH3)4] [PdCl4] + 2NH4Cl.
При дальнейшем добавлении аммиака соль Вокелена растворяется с образованием тетраамминпалладий(II) дихлорида:
[Pd(NH3)4] [PdCl4] + 4NH3 = 2 [Pd(NH3)4] Cl2.
К полученному раствору постепенно прибавляется соляная кислота: при этом выпадает светло-желтый кристаллический осадок транс-дихлородиамминопалладия(II), или палладозоамин:
[Pd(NH3)4] Cl2+ 2HCl = [Pd(NH3)2Cl2] + 2NH4Cl.
Соль мало растворима в воде и в отличие от цис-изомера более светлого цвета.
Кроме этих реакций, при добавлении аммиака протекает взаимодействие комплексов родия(III) и хлорида железа(III) с аммиаком по реакциям:
(NH4)3[RhCl6] + 3NH4OH = Rh(OH)3 + 6NH4Cl
FeCl3 + 3NH4OH = Fe(OH)3 + 3NH4Cl
Гидраты выпадают в осадок, а медь остается в растворе в виде комплекса:
CuCl2 + 4NH4OH = [Cu(NH3)4] Cl2 + 2H2O
Обработка аммиаком производится в котлах при температуре 75–85оС, осаждение палладозоамина – на холоду. В процессе осаждения палладозоамина соляной кислотой гидраты растворяются, а комплексы разрушаются и не мешают осаждению содержащего аффинируемый металл вещества. Количество соляной кислоты не должно быть очень большим, т. к. палладозоамин может снова перейти [Pd(NH3)2Cl2]+ 2HCl = (NH4)2[PdCl4].
После фильтрации и сушки палладозоамина его направляют на прокаливание:
[Pd(NH3)2Cl2] = Pd + 2HCl + NH4Cl.
4.1.3 Аффинаж иридияНа аффинаж иридия, как отмечалось выше поступают растворы и промывные воды от иридистого хлороплатината аммония. Они заливаются в резервуар, нагреваются до 95–100 оС, подкисляются азотной кислотой до прекращения бурной реакции и вскипания (расход азотной кислоты ~ 1.0–1.5 объема на 100 объемов раствора). В результате выпадает осадок гексахлороиридата(IV) аммония:
2 (NH4)3[IrCl6] + 2Cl = 2 (NH4)2[IrCl6] + 2NH4Cl
Раствор прогревается до полного осаждения соли иридия (по пробе), отстаивается, декантируется и фильтруется, осадок хлорида промывается на фильтре 8%-ным раствором аммония несколько раз. Осадок подсушивается – это технический гексахлороиридат(IV) аммония (ХИА) и вновь идет на растворение. На 100 кг (NH4)2[IrCl6] берется 100 л технической соляной кислоты плотностью 1.17 и 100 л воды. Раствор нагревается до температуры 90–95 оС, и в него вводится технический ХИА и затем небольшими порциями – азотная кислота. После растворения ХИА раствор охлаждается и фильтруется. Осадок промывается, просушивается и направляется на плавку на штейн, а раствор – на упаривание с серной кислотой до сиропообразного состояния при температуре не менее 110 оС с целью удаления удаление азотной кислоты. После этого раствор разбавляется, охлаждается и направляется на электролиз.
Необходимо подчеркнуть, что «шлиховая платина», как правило, содержит крайне малые количества родия и рутения в отличие, например, от платиновых концентратов (норильский концентрат КП‑2). Поэтому далее мы рассмотрим аффинаж металлов-спутников безотносительно к виду перерабатываемого сырья.
4.1.4 Аффинаж родияРастворы, содержащие родий, обычно упаривают для понижения кислотности и разрушения органических восстановителей, если таковые ранее использовались, при температуре 85–90 оС и в них вводится азотная кислота – примерно 1% от общего количества раствора – для разрушения избытка органических восстановителей и щавелевой кислоты, концентрация которой в исходном растворе может достигать около 8 г/л. После этого растворы упаривают до плотности 1.6–1.7 и анализируют на содержание соляной кислоты (не более 230 г./л). Далее растворы охлаждают до 45 оС, добавляют воду и вновь упаривают до плотности 1.6–1.7. Упаривание с водой ведут до тех пор, пока не получат оптимальную кислотность. Далее растворы идут на нитрование, цель которого перевести хлорокомплексы платиновых металлов в нитрокомплексы. Кроме того, операция нитрования производится для очистки растворов от неблагородных металлов: меди, железа, никеля, селена, теллура, сурьмы, свинца, которые выделяются в форме гидратов. Золото при этом восстанавливается до металла.
При нитровании реализуются следующие реакции:
NaNO2 + HCl = NaCl + HNO2
2HNO2 → NO2 + NO + H2O
2HCl + 2NaNO2 = 2NaCl + NO2 + 8NO + H2O
H3[RhCl6] + 6NaNO2 = Na3[Rh(NO2)6] + 3HCl + 3NaCl
H2[IrCl6] + 7NaNO2 = Na3[Ir(NO2)6] + 4NaCl + NO2 + 2HCl
H2[RuCl6] + 6NaNO2 = Na2[RuNO(NO2)4OH] + 4NaCl + NO2+ HCl
H2[PtCl6] + 6NaNO2 = Na2[Pt(NO2)4] + 4NaCl + 2NO2 + 2HCl
H2[PdCl4] +4NaNO2 = Na2[Pd(NO2)4] + 2HCl + 4NaCl
MeCl3+ 3NaNO2+ 3H2O = Me(OH)3↓ + 3NaCl+ 3HNO2
MeCl2 + 2NaNO2+ 2H2O = Me(OH)2↓ + 2NaCl+ 2HNO2
2HNO2 → NO2 + NO + H2O
2H[AuCl4] + 6NaNO2 → 2Au↓ + 6NaCl + 6NO2 + 2HCl.
Перед нитрованием растворы разбавляют водой до плотности 1.24. Сам процесс нитрования ведут раствором NaNO2 плотностью 1.32 в соотношении 1:1. В нагретые до 85–90 оС при интенсивном перемешивании тонкой струйкой вводится NaNO2. Затем растворы несколько разбавляются горячей водой и прогреваются при 100 оС. Избытка NaNO2 следует избегать, т. к. при последующей операции осаждения солей родия и иридия произойдет сильное разогревание растворов и выделение оксидов азота. Повышать плотность больше 1.22 нельзя, т. к. может произойти высаливание родия в гидраты. Полнота нитрования достигается при рН 3.8–4.5. По окончании нитрования растворы охлаждаются до 45 оС и оставляют до полного выделения гидратов. Осадок гидратов отмывается горячей водой (соотношение ж:т = 2:1) при интенсивном перемешивании и температуре 80 оС. Пульпа охлаждается, раствор отделяют от осадка гидратов, который обычно перерабатывают вместесо шламами медно-никелевого производства. Заметим, что если в гидратах повышено содержание родия, то их промывку ведут хлоридными родиевыми растворами. Растворы от промывки гидратов присоединяют к пронитрованным растворам. После нитрования содержание в растворах родия находится в интервале 0,5–2,5 г/л, палладия – 3–7 г./л.
Из пронитрованных растворов 25%-ным раствором NH4Cl осуществляют выделение родий-иридиевого концентрата, содержащего смешанные соли натрия и аммония АНГ (аммоний-натрий гексанитрородиат(III) и гексанитроиридат(III)) cостава (NH4)2Na [Rh(NO2)6], (NH4)2Na [Ir(NO2)6]. Избыточная концентрация хлорида аммония составляет около 8%. При осаждении идут следующие реакции:
Na3[Rh(NO2)6] + 2NH4Cl = (NH4)2Na [Rh(NO2)6]↓ + 2NaCl
Na3[Ir(NO2)6] + 2NH4Cl = (NH4)2Na [Ir(NO2)6]↓ + 2NaCl
Na2[Ru(OH) NO(NO2)4] + 2NH4Cl = (NH4)2[Ru(OH) NO(NO2)4] + 2NaCl
Na2[Pt(NO2)4] + 2NH4Cl = (NH4)2[Pt(NO2)4] + 2NaCl
Na2[Pd(NO2)4] + 2NH4Cl = (NH4)2[Pd(NO2)4] + 2NaCl.
Процесс осаждения ведут при непрерывном перемешивании. Маточные растворы после осаждения I‑го АНГ декантируются и отстаиваются, а осадок I‑го АНГ распульповывается 8%-ным раствором хлорида аммония. Пульпа проверяется на полноту отмывки от палладия, меди и никеля и направляется на высаливание. Промывные воды присоединяются к основным маточным растворам.
Высаливание – метод очистки I‑го АНГ. Его суть заключается в том, что при повышенных концентрациях иона натрия наблюдается различная растворимость нитрокомплексов платиновых металлов в водных растворах: комплексы платины, палладия и рутения почти полностью остаются в растворе, иридий остается в растворе на 90–95%, а родий высаливается в виде гексанитрородиата(III) натрия Na3[Rh(NO2)6], который называют солью НГ. Выход близок к 100%.
Цикл высаливания включает совокупность ряда операций:
1. растворение АНГ в HCl;
2. нитрование;
3. высаливание;
4. растворение НГ в воде.
5. выделение гидрокарбонатов неблагородных металлов;
6. получение АНГ высокой степени чистоты.
Указанный цикл осуществляется следующим образом. I‑ый АНГ растворяется в солянокислом растворе (на 1 л технической кислоты берут 0,5 л воды). Для этого раствор HCl нагревается до 80оС, и в него при перемешивании небольшими порциями вводится I‑ый АНГ. При растворении АНГ необходимо, чтобы каждая следующая порция соли вводилась после прекращения выделения оксидов азота предыдущей порции. Затем реакционная смесь упаривается наполовину, и раствор разбавляется горячей водой. При растворении протекают следующие реакции:
(NH4)2Na [Rh(NO2)6] + 9HCl = H3[RhCl6] + 2NH4Cl + NaCl + 3NO2 + 3NO + 3H2O
(NH4)2Na [Ir(NO2)6] + 9HCl = H3[IrCl6] + 2NH4Cl + NaCl + 3NO2 + 3NO + 3H2O.
NO2 с водой могут образовывать HNO3, следовательно, в растворе не исключено присутствие некоторого количества «царской водки», что может привести к частичному окислению Ir(III) до Ir(IV). Кроме того, возможно образование нитрозокомплексов, однако при первом и втором циклах высаливания добиваться полного разрушения нитрозокомплексов не обязательно.
Для высаливания НГ родия в раствор вводится сухой NaNO2. Количество NaNO2 – 2–3 кг на 1 кг растворенного АНГ. NaNO2 вводится в нагретый до 90 оС раствор при перемешивании. Конец операции определяется по прекращению пенообразования и началу кристаллизации соли. После нитрования растворы декантируются, прогреваются и при перемешивании вводится оставшаяся часть NaNO2, после чего растворы охлаждаются. В результате выпадает I‑ый НГ родия. Он промывается раствором NaNO2 и растворяется горячей водой. По окончании растворения и охлаждения в раствор вводится кальцинированная сода для осаждения гидрокарбонатов неблагородных металлов, которые отфильтровываются после отстаивания.
За полный цикл высаливания выводится до 90% иридия. Количество циклов определяется именно содержанием иридия. Обычно для получения в последующем металлического порошка родия, соответствующего требованиям стандарта, достаточно 3–4 циклов высаливания. Извлечение родия в каждом цикле – 99%.
Поэтому в отфильтрованные от осадков гидрокарбонатов растворы при перемешивании вводится 25%-ный раствор хлорида аммония порциями, в результате чего осаждается II‑ой АНГ родия и оставшаяся часть иридия. Если проба на полноту осаждения неудовлетворительна, то вводится дополнительное количество хлорида аммония. Осадок II‑го АНГ отделяется фильтрованием и промывается раствором NH4Cl. Маточные растворы идут на обогащение. Режимы проведения операций во всех циклах высаливания одинаковы, только расход NaNO2 на нитрование и высаливание требуется немного меньший, и растворение III‑ей и IV‑ой АНГ производится в соляной кислоте марки «чистая». Осаждение их осуществляется отфильтрованным 25%-ным раствором NH4Cl. После трех циклов очистки, если IV‑ый АНГ отвечает требованиям ГОСТ, он поступает на растворение в соляной кислоте. Раствор упаривается, фильтруется и идет на электролитическое выделение родия.
Процесс электролиза осуществляется в электролитической ванне с титановым катодом, который навешивается на медные штанги, покрытые платиновой фольгой, и анодом из родированного титана. Плотность тока – 45–50 А/дм2. В ходе процесса в ванну добавляется HCl (1:3) из расчета поддержания кислотности 80–90 г./л. Снижение содержания соляной кислоты в электролите сопровождается уменьшением электропроводности растворов и может привести к нежелательному процессу гидролиза с выделением родия в форме нерастворимого гидроксида Rh(OH)3. Электролиз длится 20–30 часов. Родий, образующийся в ходе процесса на катоде, стряхивается на дно ванны. На аноде выделяется хлор по реакции: 2Cl- – 2e → Cl2. Если в растворе будут присутствовать ионы других металлов, то все они должны восстановиться на катоде до металла, и тем самым загрязнить родий. Конец процесса электролиза определяется по цвету электролита путем его сравнения с эталонным.
По окончании электролиза катоды вынимают и промывают дистиллированной водой. Металл щеткой снимают с катода, промывают, сушат при температуре 90 оС, измельчают и просеивают. Затем порошок катодного родия загружают в платиновую лодочку с платиновой крышкой и помещают в печь, где в атмосфере водорода протекает восстан овление при температуре 1000–1100 оС в течение 2–3 часов. Водород предварительно в течение 20 минут пропускают через холодную печь. Восстановленный в указанных условиях металл охлаждается в атмосфере водорода или углекислого газа до 70–80 оС, он имеет светло-серый цвет.
Последняя операция – очистка восстановленного родия от примесей. Для этого восстановленный родий измельчается и просеивается через сито –48 меш, загружается в противень и заливается смесью HF, H2SO4 марки «х.ч.» и дистиллированной воды. Содержимое противня выпаривается досуха, охлаждается и вновь загружается той же смесью, и смесь выпаривается до появления паров SO3. Затем родий промывают дистиллированной водой до обесцвечивания промывной воды. Так удается снизить содержание кремния в металлическом родии до допустимого предела. Далее родий промывается разбавленной азотной кислотой в соотношении 1:3 в фарфоровых чашках в течение часа при 80 оС до отсутствия свинца в пробе. После проверки раствор декантируется и промывается дистиллированной водой до отсутствия в пробе хлора и свинца. Если по данным анализа в металле обнаруживается избыток железа, то он проваривается в соляной кислоте и затем тщательно отмывается.
4.1.5 Аффинаж рутенияНа аффинаж рутения поступают растворы после отделения основной массы родия и иридия в виде солей АНГ. Растворы нагреваются до 70 оС, и в них небольшими порциями вводится серная кислота. Растворы упариваются до половины объема и охлаждаются: при этом выпадает соль состава (NH4)2[RuNOCl5] – «нихра‑1». Содержимое реактора разбавляется водой до 2/3 объема, и отбирается проба на полноту осаждения рутения в виде указанной комплексной соли. Если, по данным анализа, в растворе есть рутений, то к содержимому реактора добавляется хлорид аммония до отсутствия выделяемого драгметалла в пробе.
При осаждении соединения «нихра‑1» идут следующие реакции:
2NaCl + H2SO4 = Na2SO4 + 2HCl
(NH4)2[Ru(NO) (OH) (NO2)4] + 5HCl = (NH4)2[RuNOCl5] + 5H2O + 2NO2 + 2NO
(NH4)2Na [Rh(NO2)6] + 6HCl + NH4Cl = (NH4)3[RhCl6] + 6HNO2 + NaCl
(NH4)2Na [Ir(NO2)6] + 6HCl + NH4Cl = (NH4)3[IrCl6] + 6HNO2 + NaCl
(NH4)2[Pt(NO2)4] + 4HCl = (NH4)2[PtCl4] + 4HNO2.
Образующаяся в результате превращений азотистая кислота HNO2 может частично взаимодействовать с HCl:
HNO2+ HCl =H2O + NOCl
NOCl →NO + Cl.
Свободный хлор окисляет иридий, платину, палладий до высших степеней окисления, и в результате образуются нерастворимые гексахлороиридат(IV), гексахлороплатинат(IV) и гексахлоропалладат(IV) аммония, которые загрязняют пентахлоронитрозорутенат. Поэтому соль «нихра‑1» подвергается перекристаллизации. «Нихра‑1» хорошо растворяется в воде и слабых (до 5%-ной концентрации). растворах хлорида аммония в отличие от малорастворимых гексахлороплатината(IV) и гексахлороиридата(IV) аммония. Соль рутения переходит в раствор, а соли иридия и платины остаются в осадке. Таким образом, удается отделить примеси солей платины и иридия. Если раствор рутениевой соли после фильтрации насытить хлоридом аммония до 5%, то получится более чистое соединение – «нихра‑2», которое является исходным продуктом для получения соли «нитрун».
С этой целью готовится раствор NaNO2 плотностью ~ 1.3, и в нагретый раствор вводится «нихра‑2». При этом происходит ее растворение по реакции:
(NH4)2[RuNOCl5] + 7NaNO2 + H2O = Na2[Ru(NO) (NO2)4(OH)] + 5NaCl + 3HNO2 + 2NH3.
Реакция идет очень бурно с газовыделением. Затем растворы упариваются и охлаждаются. В условиях насыщения нитритом натрия выпадает соль Na2[Ru(NO) (OH) (NO2)4]·2H2O – «нитрун». Ее растворяют в горячей воде, и из раствора производится осаждение АНГ родия и иридия сухим хлоридом аммония, после отделения которых осаждают соль «нихра‑3». Осаждение данной соли проводится чистой соляной кислотой. При этом тетранитрогидроксонитрозорутенат разрушается, и рутений переходит в пентахлоронитрозорутенат аммония. Окончание разрушения комплекса определяется по прекращению вспенивания и выделения оксидов азота. Чтобы создать необходимые условия для осаждения соли «нихра‑3», вводится сухой NH4Cl, обеспечивающий насыщение по хлориду аммония.
После промывки «нихра‑3» идет на прокаливание, которое осуществляется в электрических печах, в кварцевых тиглях, при 900 оС. Губчатый рутений отмывают водой от хлорида натрия, далее – от кремнезема смесью плавиковой и серной кислот, после чего рутений восстанавливают в трубчатой печи в атмосфере водорода.
4.1.6 Аффинаж осмияВ процессе аффинажа платиносодержащего сырья по «классической» схеме осмий повсюду следует за рутением, образуя однотипные соли (NH4)2[OsNO(NO2)4OH] и (NH4)2[OsNOCl5], разделение которых, например, фракционной кристаллизацией, не представляется возможным. Как правило, отделение осмия осуществляется на начальной стадии переработки сырья путем его отгонки в виде тетраоксида. Указанная операция проводится в перегонных аппаратах, состоящих из куба и 3–5 последовательно соединенных поглотительных емкостей, в присутствии окислителя (им может быть в том числе и «царская водка»). Поглотительные емкости, содержащие 20%-ную щелочь, охлаждают холодной водой, поскольку повышение температуры > 35 оC уменьшает улавливание OsO4. Полнота поглощения достигается добавлением в последний поглотитель, наряду с 10–15%-ным раствором NaOH, также 10–15%-ного раствора сульфида натрия.
HCl берут из расчета 1.5 кг на 1 кг перерабатываемого продукта – ее нагревают до 50–60 оС – и на 1 кг HCl вводят 0.2 – 0.3 кг HNO3. Процесс растворения в первые полчаса идет весьма энергично с выделением газов, поэтому просос воздуха вначале делают небольшим или вообще прекращают до стабилизации процесса, который продолжается в среднем 3–4 часа.
По окончании отгонки оставшийся в кубе раствор направляют на извлечение иридия, родия и других платиновых металлов, а щелочные поглотительные растворы – на выделение осмия.
При поглощении OsO4 щелочью идет следующая реакция:
OsO4 + 2NaOH = Na2[OsO4(OH)2].
Добавление в указанный раствор восстановителя – им служит тиосульфат натрия (20 мл на 1 л исходного осмиевого раствора) – сопровождается образованием осмат-иона OsO42 – по реакции:
4Na2[OsO4(OH)2] + Na2S2O3 = 4Na2OsO4 + Na2SO4 + H2SO4 + 3H2O.
Затем в полученный щелочной раствор вносят на холоду кристаллический хлорид аммония, который берется по стехиометрии реакции:
Na2OsO4 + 4NH4Cl = [OsO2(NH3)4] Cl2 + 2NaCl + 2H2O.
Образуется желтый осадок осмилтетрамминхлорида – соли Фреми, который быстро отделяют фильтрованием, промывают разбавленным (1:1 – 1:3) раствором соляной кислоты и сушат при температуре порядка 80 оС. Необходимо подчеркнуть, что в избытке хлорида аммония возможно частичное растворение соли Фреми с образованием аммиакатов осмия, а при недостатке NH4Cl осмий осаждается из раствора не полностью. Избежать потерь осмия, таким образом, можно лишь при условии соблюдения точного расчета и тщательного проведения технологических операций.
Высушенный осадок соли осмия помещают в печь и прокаливают в восстановительной атмосфере в течение нескольких часов, постепенно повышая температуру до 700–800 оC. Губчатый осмий темно-синего цвета измельчают, просеивают и проваривают в HF – для удаления кремния и в HCl – для удаления неблагородных металлов.
Рассмотрение представленных выше приемов аффинажа металлов платиновой группы, основанных на различиях в свойствах их комплексных соединений и склонности к окислительно-восстановительным превращениям, позволяет сделать вывод, что они позволяют получить металлы высокой степени чистоты, но с низким прямым извлечением в готовую продукцию. Процесс аффинажа – весьма длителен (он продолжается 3–4 недели) и характеризуется большим незавершенным производством. Отсюда вытекает потребность в разработке альтернативных технологий, в частности, экстракционных, которые базируются на результатах многочисленных исследований в области экстракции комплексов платиновых металлов различными классами экстрагентов.
Некоторые сведения по экстракции платиноидов приводятся в следующем разделе.
4.2 Экстракция комплексов платиновых металловЖидкостная экстракция – высокоэффективный процесс извлечения, концентрирования и разделения близких по свойствам элементов. Многочисленные исследования экстракции платиновых металлов всеми известными классами экстрагентов позволили сделать вывод, что для большинства комплексов наиболее высокое извлечение в органическую фазу обеспечивают соли аминов и четвертичных аммониевых оснований (ЧАО). Согласно предложенному проф. Л.М. Гиндиным еще в 60‑х годах механизму экстракции платиновых металлов аминами из кислых растворов, в органической фазе при этом образуются комплексы с N‑содержащими внешнесферными органическими лигандами.
Экстракция по механизму внешнесферного замещения уменьшается с увеличением кислотности водной фазы и возрастает в ряду аминов: первичный < вторичный < третичный. Соли ЧАО экстрагируют анионные комплексы только по механизму внешнесферного замещения, что существенно облегчает реэкстракцию и нередко обеспечивает возможность на этой стадии глубокого разделения близких по свойствам элементов.
Особенно подробно изучена экстракция из солянокислых растворов, образующихся в технологии в процессах гидрохлорирования различного сырья. Предложены схемы отделения платиновых металлов от сопутствующих им железа и цветных металлов – меди(II), никеля(II), кобальта(II) с использованием первичных, вторичных, третичных аминов, а также разделения пар платина – родий, родий – иридий, рутений – осмий и др.
Соли ЧАО – еще более эффективные экстрагенты по сравнению с солями аминов. При этом Pt(IV), Ir(IV), Ru(IV) экстрагируются значительно лучше, чем Ir(III), Ru(III), Rh(III). За счет большого различия в коэффициентах распределения можно в сильнокислых растворах осуществить разделение платины и палладия от металлов-спутников, в слабокислых – отделить платину от палладия, рутений и иридий – от родия.
Поведение комплексов платиновых металлов в водных растворах отличается многообразием химических форм, причем каждая из них характеризуется определенной величиной извлечения в органическую фазу, т.е. своим коэффициентом распределения. Последний рассчитывается как отношение равновесныйх концентраций извлекаемого комплекса, соответственно, в водной и органической фазах.
В табл. 7 приведены коэффициенты распределения DRu при экстракции различных комплексов рутения из 4 М HCl растворами три-н-октиламина (ТОА) в бензоле. В порядке уменьшения коэффициентов распределения комплексы располагаются в ряд: [RuCl6]2- > [RuNOCl5]2- > [RuCOCl5]2- Такая же последовательность соблюдается и в других растворителях, например, в CHCl3. Она совпадает с ростом дипольных моментов комплексов в том же направлении (дипольный момент комплекса складывается из дипольных моментов отдельных фрагментов; дипольный момент фрагмента Cl–Ru–Cl принимаем равным нулю, Cl–Ru–L – 2,65 D для L = NO и 3,18 D для L = CO). Следовательно, уменьшение извлечения комплексов рутения(III) и (IV) в органическую фазу находится в прямой зависимости от их гидрофильности.
Таблица 7. Коэффициенты распределения DRu при экстракции комплексов рутения (CRu=2·10-3 моль/л) из 4 М HCl растворами три-н-октиламина в бензоле
| СТОА, М | [RuCl6]2- | [RuNOCl5]2- | [RuCOCl5]2- |
| 0.0050 | 1.78 | 1.13 | 0.67 |
| 0.0075 | 4.45 | 3.90 | 2.00 |
| 0.0100 | 7.13 | 5.30 | 4.07 |
| 0.0120 | 10.0 | 9.20 | 7.08 |
| 0.0140 | 12.9 | 11.2 | 9.65 |
Коэффициенты распределения комплексов рутения(IV) при его экстракции в форме [Ru2OCl10]4- из свежеприготовленных солянокислых растворов аминами на 1–2 порядка меньше, чем для гексахлорорутената(IV), и с ростом кислотности водной фазы уменьшаются: это указывает на анионообменный характер процесса. При экстракции рутения(IV) в форме [Ru2OCl10]4 – аминами (Am) происходит его деполимеризация, и в органическую фазу извлекается моноядерный комплекс Ru(IV) по реакциям:
[Ru2OCl10]4-вод+ 4ЧAО+вод. = 2 {(ЧAО)2[RuOHCl5]}орг.
[Ru2OCl10]4-вод+ 4AmH+орг. = 2 {(AmH)2 [RuOHCl5]}орг.
Рутений(III) в форме [Ru(H2O) Cl5]2 – третичными аминами практически не экстрагируется, однако нами установлено, что он переходит в органическую фазу при экстракции первичными аминами, при этом с ростом концентрации HCl в равновесных растворах DRu не уменьшается, как это обычно имеет место в аминных экстракционных системах, а увеличивается от 0.43 в 1 М HCl до 4.0 – в 6 М HCl (СRu = 1·10-3 моль/л, Cдодециламина = 0.1 моль/л).
В отличие от пентахлороакворутената(III) комплекс рутения(III) – [RuCOCl5]2 – ведет себя при экстракции аминами из солянокислых растворов традиционно для анионных комплексов платиновых металлов. DRu возрастает по ряду аминов первичный < вторичный < третичный < соль ЧАО, причем в последнем случае коэффициент распределения приближается к 100 при концентрации экстрагента, всего в два раза превышающей концентрацию металла в водной фазе. С ростом кислотности водной фазы он падает независимо от природы амина вследствие конкурирующего влияния кислоты за экстрагент. DRu при экстракции комплекса [RuCOCl5]2 – 0,02 М раствором ди-н-октиламина в CCl4 принимает следующие значения:
| CHCl, М | 0.1 | 1 | 2 | 4 | 6 |
| DRu | 3.64 | 1.60 | 1.21 | 1.00 | 0.90 |
Карбонилхлоридные комплексы известны не только для рутения, но и для других редких платиновых металлов – родия и иридия и образуются при пропускании через солянокислые растворы их хлорокомплексов оксида углерода CO при температуре 90 оС. Характерной особенностью этих комплексов является способность растворяться не только в воде, но и в органических растворителях. Поэтому при экстракции мы наблюдаемый совокупный эффект от действия экстрагента и разбавителя. Интересно отметить, что экстракция карбонилхлоридных комплексов редких платиновых металлов также наиболее эффективно протекает при использовании аминов и солей ЧАО, причем родий в форме [Rh(CO)2Cl2]- извлекается стехиометрическим количеством экстрагентов, рутений в виде [Ru(CO)2Cl4]2- – 5–10‑кратным избытком экстрагента. Что касается иридия, то комплекс [Ir(CO)2Cl2] – даже при 50–100‑кратном избытке экстрагента в органической фазе переходит в нее на 70–80%.
Комплексы Ru(III) экстрагируются из солянокислых растворов оксидами третичных аминов, например, оксидом три-н-октиламина (ТОАО). Зависимость DRu от концентрации HCl в водной фазе при экстракции комплекса [Ru(H2O) Cl5]2 – ТОАО носит экстремальный характер с максимумом в области 8 М HCl (DRu » 14).
Одним из наиболее распространенных кислородсодержащих экстрагентов является три-н-бутилфосфат (ТБФ). Исследования показали, что зависимость коэффициентов распределения при экстракции комплексов платиновых металлов из кислых сред ТБФ от концентрации кислоты проходит через максимум, причем величина Dмет. и CHCl в экстремальной точке зависят от природы экстрагируемого иона. Как правило, наличие максимума объясняют тем, что первоначально коэффициент распределения растет в связи с протонированием кислорода экстрагента, а затем падает за счет конкурирующего действия кислоты. Такой вид зависимостей характерен для экстракции анионных комплексов по гидратно-сольватному механизму с образованием в органической фазе комплексов состава [H+(H2O)x(ТБФ)y]n[MCl6]m. Ряд экстрагируемости для комплексов платиновых металлов имеет вид:
Au(III) > Pt(IV) > Pd(II) > Ir(IV) > Ir(III).
Извлечение возрастает по ряду ацидолигандов Cl- < Br- < I-, что обусловлено уменьшением гидратации и увеличением устойчивости комплексов. В ряду разбавителей н-гексан > толуол > 1.2‑дихлорэтан > хлороформ экстрагируемость комплексов три-н-бутилфосфатом обычно уменьшается вследствие взаимодействия экстрагента и разбавителя. Следйет отметить, что в области 6 М HCl извлечение платины(IV) намного выше, чем палладия(II). Для 90%-ного раствора ТБФ в гексане коэффициент разделения платины и палладия равен 1250, в толуоле он достигает значения 1640, т.е. возможно разделение этих металлов за одну ступень экстракции.
По мере замены заместителей R-O- в молекуле ТБФ на алкильные R – растет электронная плотность на фосфорильном кислороде, ответственном за комплексообразование, и, соответственно, возрастает экстракционная способность реагентов. В зависимости от строения углеводородных радикалов и длины углеродной цепи коэффициенты распределения изменяются следующим образом (показано на примере экстракции Pd(II) – CPd = 5·10-3 моль/л – из 3 М HCl 0.1 М раствором фосфиноксида в толуоле или бензоле):
Фосфиноксид DPd
| Тригексил | 1.07 |
| Дигексилоктил | 0.63 |
| Тригексил | 0.888 |
| Диоктилгексил | 1.03 |
| Триоктил | 0.75 |
| Тринонил | 1.00 |
В отличие от комплексов других платиновых металлов палладий(II) особенно эффективно извлекается серосодержащими экстрагентами – сульфидами формулой R2S. Обычно товарные продукты такого рода представляют собой смеси сульфидов в углеводородах. В процессе экстракции происходит отделение палладия от рутения, родия (коэффициенты разделения составляют 105 ¸ 106). Наиболее трудно отделяется серебро в азотнокислых растворах, поэтому целесообразен для достижения этой цели переход к солянокислым системам.
При экстракции органическими сульфидами и сульфоксидами (R2SO) последовательность перехода комплексов благородных металлов из водной фазы в органическую представляется так:
Au(III) > Pd(II) > Pt(II) >> Rh(III) > Ru > Ir(III).
Высокая избирательность сульфидов по отношению к иону Pd(II) обусловлена тем, что в отличие от нейтральных кислородсодержащих экстрагентов они с большим трудом протонируются и не извлекают анионные комплексы платиновых металлов. Экстракция протекает за счет непосредственной координации молекул экстрагента к извлекаемому иону. Наличие кислорода в сульфоксидах и сульфонах приводит к снижению электронной плотности на атоме серы, в результате экстракционная способность от сульфидов к сульфоксидам снижается. В этом случае не исключено протонирование кислорода и экстракция анионных комплексов.
В технологии аффинажа приходится сталкиваться с нитритными растворами, в которых платиновые металлы присутствуют в форме нитрокомплексов. Они практически не экстрагируются аминами из нейтральных сред, однако при подкислении растворов степень извлечения в органическую фазу возрастает. Так, пятистадийная экстракция платины(II), палладия(II), иридия(III) из нитритных растворов три-н-октиламином при рН 1–2 обеспечивает практически полное (> 99.9%) их извлечение. Характерно, что рутений экстрагируется из нитритных сред значительно лучше платины и палладия: при рН 3.5 – 4.2 DRu составляет 33.1, в то время как DPt = 0.87, DPd = 1.95.
В процессе экстракции нитрокомплексов платиновых металлов из нитритных растворов солями ЧАО они извлекаются в органическую фазу без разрушения внутренней координационной сферы. Установлено, что нитрокомплексы платины(II) и палладия(II) экстрагируются солями ЧАО в органическую фазу с высокими коэффициентами распределения даже из растворов, содержащих 40–60% NаNO2. Так, при экстракции комплексов [Pd(NO2)4]2- и [Pt(NO2)4]2 – (CМ = 0,001 моль/л) из 50%-ных по нитриту натрия растворов три-н-октилбензиламмонийхлоридом (ТОБАХ) в CCl4 (CТОБАХ = 0,003 М) получены DPd = 3.89 и DPt = 6.75. В тех же условиях ни родий(III), ни иридий(III) не экстрагируются ТОБАХ в CCl4: даже при экстракции [Rh(NO2)6]3- из раствора, содержащего 10% NаNO2: коэффициент распределения не превышает 0,1. В порядке уменьшения коэффициентов распределения нитрокомплексы располагаются следующим образом:
[Pt(NO2)4]2- > [Pd(NO2)4]2- > [Ir(NO2)6]3- > [Rh(NO2)6]3-.
Найдены условия количественного извлечения рутения и осмия в форме нитрозонитрокомплексов наиболее распространенными экстрагентами.
4.3 Сорбция комплексов платиновых металловПараллельно с развитием экстракционных методов велись поиски сорбентов, селективных к платиновым металлам. Сорбцию выгодно отличает от жидкостной экстракции технологичность и быстрота в исполнении, а также возможность работы с многокомпонентными природными и промышленными материалами: рудами, горными породами, продуктами аффинажного производства, в том числе бедными по содержанию металлов платиновой группы. Наиболее часто сорбционное концентрирование встречается в аналитической практике.
Сорбция – процесс поглощения газов, паров, растворенных веществ (сорбатов) твердыми и жидкими поглотителями (сорбентами). Различают следующие виды сорбции: адсорбция – поглощение веществ на поверхности твердого или жидкого тела; абсорбция – поглощение газов, паров или растворенных веществ во всем объеме твердой или жидкой фазы; хемосорбция – поглощение веществ твердыми или жидкими сорбентами с образованием химических соединений; капиллярная конденсация – образование жидкой фазы в порах и капиллярах твердого сорбента при поглощении паров вещества. На практике различные виды сорбции, как правило, сочетаются друг с другом. При концентрировании следовых количеств ценных компонентов наиболее часто встречается адсорбция и хемосорбция, последняя обычно происходит путем ионного обмена либо комплексообразования.
По способу осуществления сорбционные процессы делятся на статические и динамические. Главными характеристиками сорбционных процессов являются: коэффициент распределения (Kраспр) и степень извлечения сорбируемого вещества (R), выражения для которых приведены ниже:
qСТ
![]() K распр =
K распр =
QАВTcСж,
где qСT – количество поглощенного вещества С в твердой фазе;
QАВT-количество твердой фазы (сорбента);
cСж-концентрация вещества С в жидкой фазе.
(Сначж - Сконж)×100
![]() R=
R=
Сначж
Сначж - концентрация сорбируемого вещества в исходном растворе;
Сконж – концентрация сорбируемого вещества в растворе после сорбции.
В качестве обобщенной характеристики сорбентов обычно используют удельную поверхность – площадь поверхности 1 г сорбента. Отметим также, что для сорбентов с определенным типом взаимодействующих с молекулами сорбата функциональных групп критерием служит концентрация функциональных групп на поверхности. Ионообменные сорбенты на основе полимерных органических матриц характеризуются величиной сорбционной емкости, т.е. числом молей вещества, поглощаемых 1 г ионообменника.
Применение сорбционных методов концентрирования и разделения платиновых металлов исторически связано с ионным обменом. Первоначально в качестве ионообменников использовали алюмосиликаты и цеолиты. Активное применение ионообменных методов для разделения близких по свойствам элементов началось 30–40 лет назад, в связи с синтезом в нашей стране и за рубежом большого числа органических соединений – ионитов, обладающих сильноосновными и сильнокислотными свойствами. Это хорошо известные сорбенты АВ‑17, Дауэкс‑1, КУ‑2, ЭДЭ‑10П, Дауэкс‑50 и др. Использование указанных ионитов позволило, в частности, решить задачу отделения следов платиновых металлов от значительно преобладающих количеств цветных, редкоземельных, щелочных и щелочноземельных металлов, однако трудность десорбции платиновых металлов с данных сорбентов помешала их широкому распространению.
Следующим этапом в развитии сорбционных процессов (60–80‑е годы) стало применение слабокислотных катионитов, слабоосновных анионитов и амфолитов, главными достоинствами которых является высокая селективность и возможность регулирования гранулометрического состава. Благодаря им, удалось решить задачу концентрирования платиновых металлов на фоне ионов меди, никеля, железа, кобальта, присутствующих в рудном платиносодержащем сырье.
Нельзя не отметить, что органические ионообменники имеют существенные недостатки, как то: набухаемость, невысокая механическая прочность, практически невозможность десорбции платиновых металлов с поверхности сорбента. Перечисленных недостатков лишены неорганические ионообменники – к ним относятся, например, оксигидратные сорбенты R2O3·nH2O, где R – Fe, Al, Zr, Y, Sm, Eu, отличающиеся, напротив, ненабухаемостью в растворителях, механической прочностью, термостабильностью и радиационной устойчивостью. Превосходя сорбенты на основе органической полимерной матрицы также и по скорости сорбции, неорганические сорбенты, безусловно, уступают им в эффективности и селективности по отношению к платиновым металлам.
Полезные кинетические свойства неорганических и селективные – полимерных матриц объединяют в себе коплексообразующие ионообменники на основе силикагелей. В конце 80‑х годов интенсивное развитие получила область синтеза и применения химически модифицированных кремнеземов (ХМК). Механические и массообменные характеристики ХМК определяются жестким широкопористым каркасом носителя, а ионообменные и комплексообразующие – природой закрепленных функциональных групп.
Ионообменные свойства сорбентов определяются привитыми функциональными группами. В зависимости от знака заряда сорбируемых ионов они подразделяются на катиониты, аниониты и амфолиты (амфотерные иониты) Применительно к платиновым металлам принципиальной разницы между слабоосновными анионитами, амфолитами и комплексообразующими сорбентами нет. Сорбенты содержат функциональные группы, способные как к ионному, так и к координационному взаимодействию. Так же условным является выделение в самостоятельный класс сорбентов волокнистой структуры, если к ним подходить с позоций взаимодействия комплексов с функциональными группами сорбентов. Различие состоит в том, что сорбенты волокнистой структуры имеют значительно более развитую поверхность. Они построены из линейных (или разветвленных) макромолекул и обладают лучшей способностью к набуханию. Гранулированные сорбенты – чаще всего полимеры пространственного строения. Волокнистые сорбенты выгодно отличаются кинетическими и емкостными характеристиками, а разнообразная геометрическая форма (волокно, пряжа, ткань) позволяет осуществлять процесс сорбции в различном конструктивном оформлении.
В табл. 8 представлены результаты ионообменной сорбции хлорокомплексов Pt(II), Pt(IV), Pd(II), Pd(IV), Os(IV), Ir(III), Ir(IV) из солянокислых растворов на сильноосновной анионообменной смоле Дауэкс‑1. Видно, что с уменьшением концентрации соляной кислоты значения коэффициентов распределения увеличиваются, что характерно для анионообменного механизма сорбции. Обращает на себя внимание существенное отличие коэффициентов распределения при сорбции Ir(III) и Ir(IV) (Краспр Ir(III): Краспр Ir(IV) = 1:1000), которое, без сомнения, связано с различным зарядом сорбируемых ионов.
 Таблица 8. Коэффициенты распределения при сорбции платиновых металлов (Смет ³ 2 ммоль/л) из солянокислых растворов на смоле Дауэкс‑1
Таблица 8. Коэффициенты распределения при сорбции платиновых металлов (Смет ³ 2 ммоль/л) из солянокислых растворов на смоле Дауэкс‑1 | СHCl, М Комплекс | 1,2 | 3 | 6 | 9 |
| [PtCl4]2- | 104 | 5000 | 2800 | 710 |
| [PtCl6]2- | 104 | 3600 | 1900 | 870 |
| [PdCl4]2- | 1300 | 500 | 120 | 50 |
| [PdCl6]2- | 1600 | 350 | 80 | 55 |
| [OsCl6]2- | 104 | 104 | 4800 | 3700 |
| [IrCl6]3- | 16 | 7,7 | 2,3 | 1,5 |
| [IrCl6]2- | 104 | 7000 | 2600 | 1500 |
Селективность комплексообразующих сорбентов по отношению к ионам металлов либо комплексным ионам обусловлена природой функциональных групп, закрепленных на полимерной матрице. Кроме того, на взаимодействие с сорбируемыми ионами, сильно влияет полимерная матрица. Наибольшую избирательность к металлам платиновой группы проявляют комплексообразующие сорбенты, содержащие в качестве функциональных групп атомы азота и серы, например, алифатические, ароматические, гетероциклические амины, тиоамиды и т.п.
Сорбенты на основе сополимеров стирола, акрилонитрила, метакрилата, сополимеров целлюлозы, содержащие в качестве функциональных групп гетероциклические амины, получили наибольшее распространение. Они имеют товарный знак ПОЛИОРГС.
ПОЛИОРГС'ы химически устойчивы в водной среде в широком диапазоне кислотности, а также при нагревании растворов до 100 оС. Применяются в виде гранул, порошков и волокон. К несомненным достоинствам этих сорбентов следует отнести высокую емкость по платиновым металлам, что подтверждают обобщенные данные, приведенные в табл. 9. Сорбционная емкость зависит от природы матрицы. Например, при переходе от аминополистирола (ПОЛИОРГС V) к поликонденсату бензимидазола и меламина (ПОЛИОРГС XXI) емкость сорбента по иридию возрастает в 116 раз. Селективность возрастает в ряду пиразол < бензимидазол < имидазол. Полиоргс VI – волокнистый сорбент на основе поливинилового карбоцепного волокна с 3 (5) – метилпиразолом. Он способен селективно извлекать платиновые металлы в присутствии 106-кратного избытка Cu(II), Ni(II), Fe(III). Сорбцию следует вести при кипячении раствора в течение часа. При комнатной температуре из 1М HCl Ir (Ru, Rh) не сорбируются. ПОЛИОРГС X – волокнистый сорбент, содержащий аминогруппы, устойчив в кислых и щелочных средах. В 2М HCl сорбционная емкость по иридию составляет 90 мг/г. Она сохраняется в присутствии 5*106-кратных избытков Cu(II), Cо(II), Ni(II), Fe(III).
По скорости достижения равновесия на всех сорбентах класса ПОЛИОРГС хлорокомплексы металлов платиновой группы можно расположить в ряд: Pd(II) > Pt(IV)> Ir(IV) > Rh(III) > Ru(IV) >> Ru(III).
Десорбция платиновых металлов с сорбентов ПОЛИОРГС затруднена. Например, с сорбентов ПОЛИОРГС IV, V и VI осуществить десорбцию практически не удается даже при использовании сильных комплексообразующих соединений (чаще всего – тиомочевины), что является их существенным недостатком. Однако поскольку сорбционное концентрирование на ПОЛИОРГС'ах используется преимущественно аналитических целях, десорбцией можно пренебречь.
Таблица 9. Сорбционная емкость сорбентов ПОЛИОРГС| Сорбент | Среда | Сорбционная емкость, ммоль/г | ||||
| Pt(IV) | Pd(II) | Rh(III) | Ir(IV) | Ru(IV) | ||
| ПОЛИОРГС V | 2М HCl | – | 0,17 | 0,12 | 0,06 | 0,12 |
| ПОЛИОРГС XI | 1М HCl | 1,52 | 2,82 | 2,72 | 1,46 | 1,78 |
| ПОЛИОРГС XV | 0,1М HCl | 0,44 | 0,82 | 1,28 | 0,29 | – |
| ПОЛИОРГС XVII | 0,1М HCl | 0,74 | 1,13 | 0,56 | 0,17 | – |
| ПОЛИОРГС XXI | 0,1М HCl | 4 | 3,28 | 8,26 | 6,97 | – |
| ПОЛИОРГС XХVI | 1М HCl | 1,28 | 2,35 | 1,94 | 2,26 | 2,9 |
| ПОЛИОРГС XХVII | 0,1М HCl | 1,79 | 3,6 | 1,94 | 1,30 | 1,94 |
Процесс сорбции может быть рекомендован для извлечения ценных компонентов из маточных и сбросных растворов. Имеющаяся обширная информация по этому вопросу позволит всегда подобрать нужный сорбент в зависимости от конкретно поставленной задачи.
[1] гексагональная плотноупакованная
[2] гранецентрированная кубическая
Похожие работы

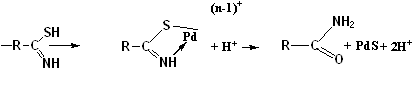

... . При уменьшении концентрации соляной кислоты величина десорбции падает. Выводы 1. Волокна ЦМ-А2, Мтилон-Т и ВАГ можно использовать для сорбции палладия из хлоридных растворов. 2. По изотермам сорбции палладия волокнами ЦМ-А2, Мтилон-Т и ВАГ установлено, что на волокне Мтилон-Т сорбционная емкость больше, чем на волокне ЦМ-А2 и ВАГ и составляет 2,92 ± 0,1 мг-экв/г, 2,13 ± 0,1 мг-экв/г и ...
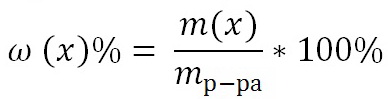
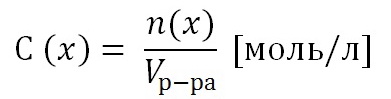
... с кислородом, восстановлением - отнятие кислорода. С введением в химию электронных представлений понятие окислительно-восстановительных реакций было распространено на реакции, в которых кислород не участвует. В неорганической химии окислительно-восстановительные реакции (ОВР) формально могут рассматриваться как перемещение электронов от атома одного реагента (восстановителя) к атому другого ( ...
... СССР, носящий ныне имя академика Н.С. Курнакова. В этом институте под руководством выдающихся ученых – Л.А. Чугаева, Н.С. Курнакова, И.И. Черняева – были выполнены многочисленные исследования по химии и технологии платины и других благородных металлов. Результаты этих исследований стали научной основой нынешней платиновой промышленности Советского Союза. Получение платины Казалось бы, раз ...
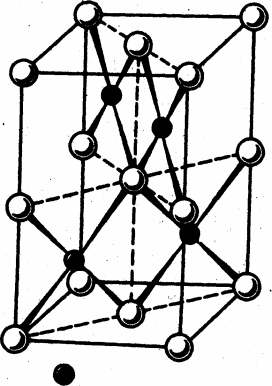
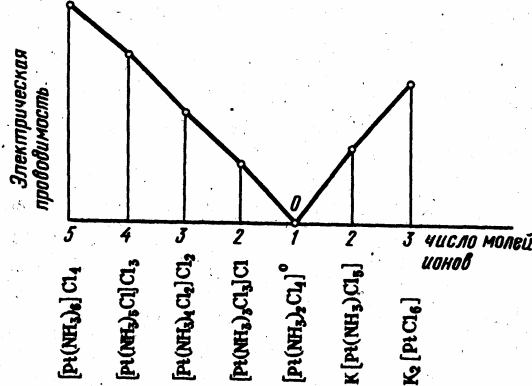
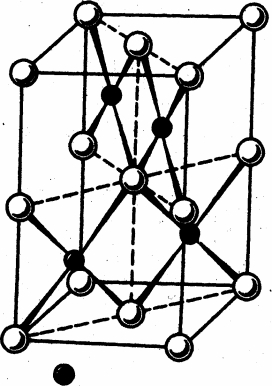
... ] — кристаллическое вещество оранжевого цвета, устойчиво при 20° С, в вакууме возгоняется без разложения. Синтез Xe[PtF6] ярился началом широких исследований, приведших к получению соединений благородных газов. Заключение Химия платины очень объемна, сложна и интересна. Пожалуй, наиболее общим свойством ее соединений является узкий температурный интервал их стабильности, связанный с высоким ...
0 комментариев